
Primary myelofibrosis with concurrent CALR and MPL mutations:A case report
2020-04-08 06:07
World Journal of Clinical Cases 2020年22期
Feng-Ping Zhou,Hua-Ping Du,Jin Zhang,Department of Hematology,Sir Run Run Shaw Hospital,Zhejiang University School of Medicine,Hangzhou 310016,Zhejiang Province,China
Cheng-Cheng Wang,Shan-Bo Cao,Acornmed Biotechnology Co.,Ltd.,Beijing 100176,China
Abstract BACKGROUND Primary myelofibrosis (PMF) is a myeloproliferative neoplasm (MPN)characterized by recurrent mutations in the JAK2,CALR,and MPL genes. The CALR and MPL co-mutation is very rare. To our knowledge,no more than five cases have been reported. Here,we report a case of PMF in which a CALR and MPL co-mutation was detected by next-generation sequencing (NGS) technology,and a literature review was performed.CASE SUMMARY A 73-year-old woman was admitted to our hospital in 2018 due to abdominal distension. The patient had splenomegaly,lymphadenopathy,leukopenia,anemia,and immature granulocytes in peripheral blood. There were dacrocytes and atypical megakaryocytes in bone marrow,and megakaryocytic proliferation was very active,accompanied by reticulin fibrosis grade 2. By NGS analysis of the bone marrow sample,we detected mutations in CALR,MPL,and PIK3RI,while JAK2 V617F and BCR-ABL were negative. Therefore,the patient was diagnosed with PMF and received oral ruxolitinib. However,the spleen and hematologic responses were poor. We review the literature,analyze previous reports of the mutation sites in our patient and differences between our patient and other reported cases of co-mutated CALR and MPL genes,and discuss the reason why the CALR and MPL co-mutations are rare and possible mechanisms and their impact on the prognosis of patients.CONCLUSION CALR and MPL mutations can be concurrent in MPN,but they are rare. The use of NGS may help to identify more patients with co-mutated CALR and MPL genes.This will help to further explore the mechanism and its impact on these patients to develop appropriate treatment strategies.
Key Words:Primary myelofibrosis; CALR; MPL; Co-mutation; Next-generation sequencing; Case report
INTRODUCTION
Primary myelofibrosis (PMF) is a myeloproliferative neoplasm (MPN) characterized by recurrent mutations in theJAK2,CALR,andMPLgenes. Although these mutations were initially reported to be mutually exclusive in MPN,several studies have reported the presence ofJAK2-CALRorJAK2-MPLco-mutations in PMF patients[1-4],andCALRandMPLco-mutation cases are still very rarely reported. With the increasingly wide application of next-generation sequencing (NGS) technology,rare myelofibrosis cases with co-mutatedCALRandMPLgenes are increasingly being detected. Here,we report a patient with PMF,in which co-mutatedCALRandMPLgenes were detected.
CASE PRESENTATION
Chief complaints
Abdominal distension for 2 mo.
History of present illness
A 73-year-old woman was admitted to Sir Run Run Shaw Hospital,Zhejiang University School of Medicine in 2018 due to abdominal distension for 2 mo,especially after eating with no complaints of abdominal pain,diarrhea,vomiting,or reduced anal exhaust and defecation.
History of past illness
The patient had a history of a left kidney cyst.
Personal and family history
She denied any other specific personal or family history of other diseases.
Physical examination
The patient’s conjunctiva and skin were pale,and the sclera was not yellow. Slightly swollen lymph nodes could be felt in the neck and armpit. The abdomen looked a little distended and felt soft,with no tenderness; the spleen under the ribs could be felt,and the texture was slightly hard. The rest of the abdomen did not have any obvious masses. The bowels sounded normal.
Laboratory examinations
Peripheral blood tests demonstrated a leukocyte count of 2.9 × 109/L,platelet count of 126 × 109/L,and hemoglobin of 87 g/L,and a smear revealed immature granulocytes of 4%. Liver function tests showed normal levels of transaminases,alkaline phosphatase,gamma-glutamyl transferase,total bilirubin,direct bilirubin,indirect bilirubin,serum creatinine,and potassium.Lactate dehydrogenase was 570 IU/L,and total cholesterol was 2.6 mmol/L.
Imaging examinations
An abdominal computed tomography scan showed that the spleen was enlarged and uniform (Figure 1),with a left kidney cyst. Abdominal ultrasound showed that the spleen was enlarged,with a thickness of 5.6 cm and a length of 16.1 cm.
Pathological examination
Bone marrow aspiration revealed erythroid and megakaryocytic hyperplasia;dacrocytes were easily observed,and megakaryocytes had atypia (Figure 2). Bone marrow biopsy showed megakaryocytic proliferation and atypia,accompanied by reticulin fibrosis grade 2 (Figure 3).
Examination for gene mutations
The bone marrow samples were sequenced by NGS,which showed mutations inCALR,MPL,andPIK3RI(Table 1),whileJAK2 V617FandBCR-ABLwere negative.
FINAL DIAGNOSIS
The patient was ultimately diagnosed with PMF (International Prognostic Scoring System score = 3,high risk).
TREATMENT
The patient received oral ruxolitinib 15 mg twice per day.
OUTCOME AND FOLLOW-UP
Half a year later,the spleen response was poor,and the size of the spleen increased from 16.1 × 5.6 cm to 17.7 × 6.6 cm. Anemia was progressively aggravated in the meantime and developed to blood transfusion dependence,indicating that PMF with co-mutatedMPLandCALRgenes cannot benefit from ruxolitinib.
DISCUSSION
Most PMF patients carryJAK2,MPL,orCALRdriver mutations[5]. Mutations inMPLandCALRare drivers in the pathogenesis of MPNs,and mutantCALRcan bind withMPLto activate the JAK-STAT signaling pathway. Previous studies have shown thatJAK2,MPL,andCALRare mutually exclusively mutated in MPN patients[6,7]. However,studies have also reported the presence ofJAK2-CALRorJAK2-MPLco-mutations in PMF patients[1-4],and cases ofCALRandMPLco-mutations are still very rarely reported. Here,we report the case of a PMF patient withCALR-p.364fs andMPLp.X636W mutations. TheCALR-p.364fs mutation is usually detected in essential thrombocytosis (ET) and PMF patients,while theMPL-p.X636W mutation is not a common mutation and has only been reported in refractory anemia with ring sideroblast patients[8,9]. Furthermore,we also detected a low-burdenPIK3R1mutation in this case. ThePIK3R1gene belongs to the Akt signaling pathway and plays a direct role in regulating cell survival,growth,and differentiation. MutatedPIK3R1can cause aberrant PI3K signaling,and the p.R228fs mutation site has already been reported in acute lymphocytic leukemia patients[10,11]. To the best of our knowledge,this is the first report of aCALRandMPLdouble mutant PMF patient from East Asia.
Earlier research reports show thatCALRandMPLmutations are mutually exclusive in MPN patients. This case demonstrated thatCALRandMPLmutations can coexist in MPN patients. Three MPN cases with co-mutatedCALRandMPLgenes have already been reported since 2017[12-14]. Partoucheet al[12]reported that a post-ET PMF patient carried type ICALRandMPL W515Rmutations,with an variant allele frequency (VAF) of 59.1% and 31.7%,respectively. Tashkandiet al[13]reported a post-ET PMF patient carrying aCALR(p.L367Tfs*46) mutation at an allele frequency of 47% and twoMPL(p.S505C and p.W515L) mutations at a lower allele frequency of 4%. Bernalet al[14]detected aCALR(p.L367fs*48) mutation at an allele frequency of 30% and aMPL(p.Trip515L) mutation at an allele frequency of 11.75% in an ET patient. In contrast to these reported cases,we identified an uncommon mutation inMPL(p.X636W) in PMF patients,and its VAF was higher than that of theCALRmutation in our patient.
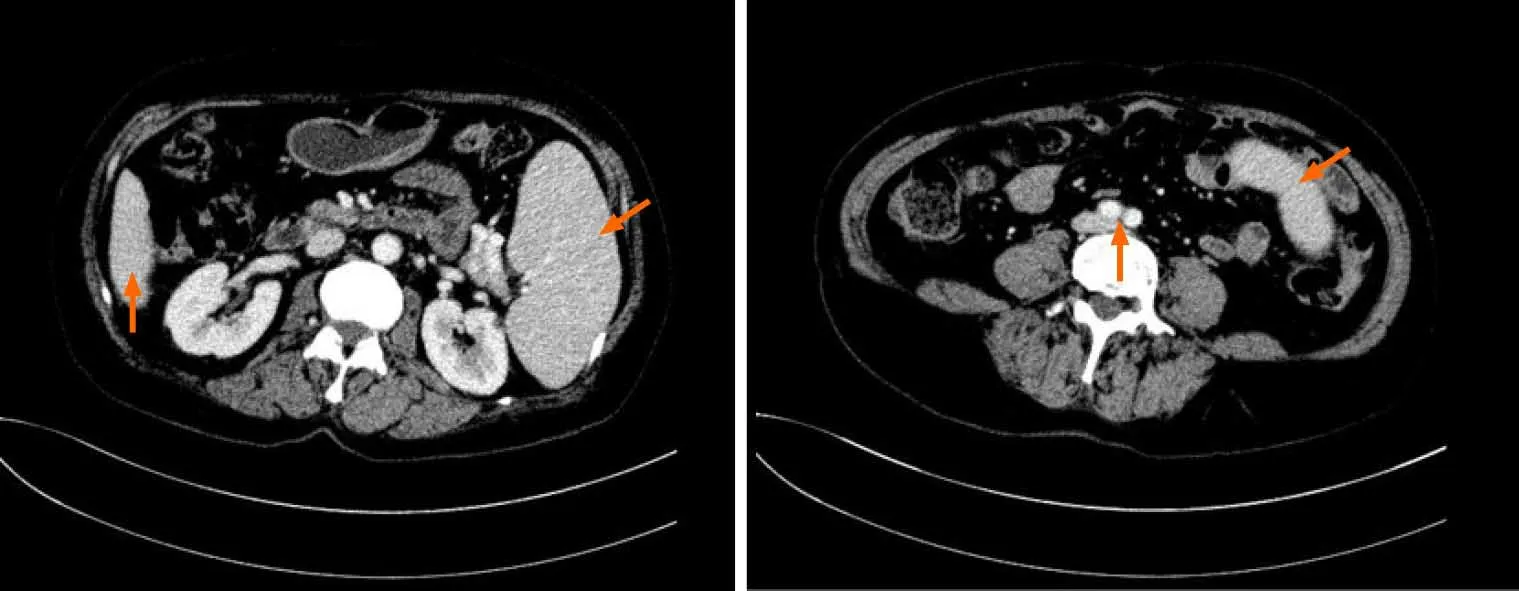
Figure 1 Computed tomography images showing splenomegaly. A and B:Abdominal computed tomography images showing that the inferior margin of the spleen was lower than that of the liver (A) and reached the level of abdominal aortic bifurcation (B).
Patients with concurrentCALRandMPLmutations are truly rare in the clinic. In PMF patients,the frequency ofCALR(20%-25%) andMPL(7%) gene mutations is low[1,6,7]. Before 2017,there were no case reports aboutCALRandMPLco-mutations.Currently,a few MPN patients have been detected,which is inseparable from the progress of molecular testing technology. From previous cases,we can see that in MPN patients,aMPLmutation VAF is usually low[2,3,15],and some uncommon mutations exist. Traditional Sanger sequencing may miss these mutations because of its test range and sensitivity[7,13-15]. High-throughput sequencing may cover the full loci,and the detection VAF limit was lowered to 1%. Therefore,MPN patients with concurrentCALRandMPLmutations can be accurately detected.
TheMPLmutation may have been a molecular event independent of theCALRdeletion or a secondary event in theCALR-containing clone. Some researchers have proven that theCALRgene mutation is an early event during disease development,andMPLis a passenger event[2,13]. It is speculated that the mutatedCALRgene can trigger genome instability,which then causes mutations in theMPLgene[13]. This is a possible explanation for whyCALRandMPLcan co-mutate. However,in our case,theMPLmutation was improbable as a secondary event because its VAF was more than 50%,indicating the possibility of a germline mutation. Some research has already proven that MPN patients can carry germline MPN mutations[16-18]. The co-mutation mechanism is not clear and still needs more investigations.
Gene mutations can predict prognosis in PMF patients.CALRmutations indicate a good prognosis in ET and PMF patients[19,20]. Compared toJAK2or triple-negative patients,CALRmutation-positive patients have a longer overall survival and low risk of thrombogenesis[1,7,19,21]. Patients who harbor theMPL W515L/Kmutation have a moderate prognosis and higher risk of thrombogenesis thanCALRmutation-positive patients[19]. The prognosis of patients withMPLandCALRco-mutations is still unknown due to the limited number of patients,but it seems that these patients have more aggressive progression based on our patient and other reported cases[12,13]. Our patient’s disease progression was rapid. In Partouche’s and Tashkandi’s patients,theCALRmutation was a pre-existing clone in the ET stage,and theMPLmutation was acquired when the disease developed into PMF. Disease acceleration may be associated withMPL-mutated clones. Because of the lack of patients with coexisting mutations,further investigations in large cohorts are needed to better quantify the presence and clinical significance.
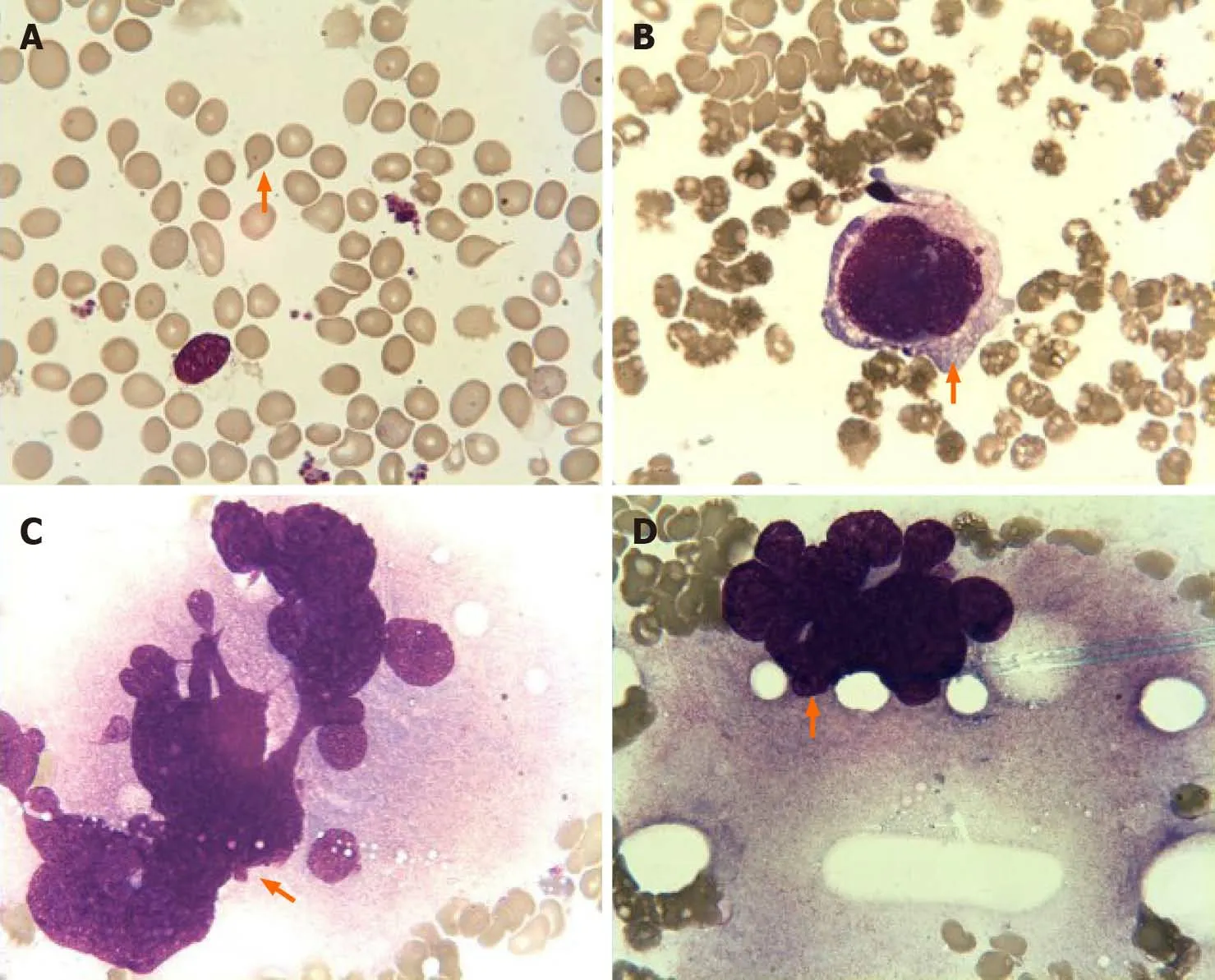
Figure 2 Bone marrow aspiration findings. A:Dacrocytes were easily observed; B-D:Images showing the presence of megakaryocyte atypia,including abnormal nuclear-cytoplasmic ratios,malformed nucleus (B),abnormal chromatin clumping with hyperchromatic nucleus (C),and extensive lobulation of the nucleus(D).

Figure 3 Bone marrow biopsy findings.A:Hematoxylin and eosin staining showed megakaryocytic proliferation and clustered distribution (magnification,200×); B:Gomori staining showed diffuse and dense increase in reticulin fibres (magnification,200 ×).
CONCLUSION
In conclusion,we have described a rare case of PMF with concurrentCALRandMPLmutations. This demonstrates thatCALRandMPLmutations are not mutually exclusive in MPN. The use of NGS may help to identify more patients withCALRandMPLco-mutations. This will help to further explore the mechanism and its impact on these patients to develop appropriate treatment strategies.
 World Journal of Clinical Cases2020年22期
World Journal of Clinical Cases2020年22期
- World Journal of Clinical Cases的其它文章
- COVID-19:A review of what radiologists need to know
- Holistic care model of time-sharing management for severe and critical COVID-19 patients
- Bioequivalence of two esomeprazole magnesium enteric-coated formulations in healthy Chinese subjects
- Osteoprotegerin,interleukin and hepatocyte growth factor for prediction of diabetes and hypertension in the third trimester of pregnancy
- High serum lactate dehydrogenase and dyspnea:Positive predictors of adverse outcome in critical COVID-19 patients in Yichang
- Risk factors analysis of prognosis of adult acute severe myocarditis