
Hemophagocytic lymphohistiocytosis caused by STAT1 gain-offunction mutation is not driven by interferon-γ: A case report
2020-04-07 06:17NanLiuFenYingZhaoXiaoJunXu
World Journal of Clinical Cases 2020年23期
Nan Liu, Fen-Ying Zhao, Xiao-Jun Xu
Nan Liu, Fen-Ying Zhao, Xiao-Jun Xu, Department of Hematology-oncology, Children’s Hospital,Zhejiang University School of Medicine, Hangzhou 310003, Zhejiang Province, China
Abstract BACKGROUND Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory syndrome caused by many genetic defects. STAT1 is a DNAbinding factor that regulates gene transcription. HLH caused by STAT1 gain-offunction (GOF) mutations has rarely been reported and its clinical manifestations and mechanisms are not clearly defined.CASE SUMMARY A 2-year-old boy presented to our hospital with recurrent fever for > 20 d. The patient had a personal history of persistent oral candidiasis and inoculation site infection during the past 2 years. Hepatosplenomegaly was noted. Complete blood cell count showed severe anemia, thrombocytopenia and neutropenia.Other laboratory tests showed liver dysfunction, hypertriglyceridemia and decreased fibrinogen. Hemophagocytosis was found in the bone marrow. Chest computed tomography showed a cavitary lesion. Tests for fungal infection were positive. Serum T helper (Th) 1/Th2 cytokine determination demonstrated moderately elevated levels of interleukin (IL)-6 and IL-10 with normal interferon(IFN)-γ concentration. Mycobacterium bovis was identified in bronchoalveolar lavage fluid by polymerase chain reaction. Genetic testing identified a heterozygous mutation of c.1154C>T causing a T385M amino acid substitution in STAT1. Despite antibacterial and antifungal therapy, the febrile disease was not controlled. The signs of HLH were relieved after HLH-94 protocol administration,except fever. Fever was not resolved until he received anti-tuberculosis therapy.Hematopoietic stem cell transplantation was refused and the patient died six months later due to severe pneumonia.CONCLUSION Patients with STAT1 GOF mutation have broad clinical manifestations and may develop HLH. This form of HLH presents with normal IFN-γ level without cytokine storm.
Key Words: Hemophagocytic lymphohistiocytosis; Signal transducer and activator of transcription 1; Gain-of-function; Interferon gamma; Mycobacterial disease; Case report
INTRODUCTION
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory syndrome that is characterized by persistent fever, hepatosplenomegaly, cytopenia,coagulopathy and impaired liver function. It comprises primary and secondary forms.The former occurs in the presence of an underlying predisposing genetic defect, while the latter presents as a complication of infection, malignancy, or autoimmune diseases without such genetic defect. These defects may be associated with lymphocyte cytotoxic defects, abnormalities of inflammasome activation, or disorders affecting inflammation. Next-generation sequencing is widely used in patients with HLH to investigate the underlying genetic defects, and an increasing number of novel genetic mutations are reported to contribute to primary HLH, which is important for identifying patients prone to HLH and to make proper clinical decisions.
STAT1 mediates the actions of many cytokines involved in mounting innate and adaptive immune responses to viruses and intracellular bacteria[1]. Signals from the interferon (IFN)-γ and type I IFN receptors are transducedviaa Janus kinase(JAK)/STAT pathway that uses STAT1[2]. STAT1 is one of the nine genes that can cause Mendelian susceptibility to mycobacterial disease, which is a rare condition characterized by predisposition to clinical diseases caused by weakly virulent mycobacteria with impaired production of or response to IFN-γ[3]. Here, we report a patient with gain-of-function (GOF) STAT1 mutation who developed pulmonary aspergillosis, mycobacterial disease and HLH. To the best of our knowledge, this is the first case report of GOF STAT1 mutation leading to HLH under conditions without previous primary immunodeficiency disease, which presents some distinct features in cytokine profile and treatment response compared with other primary forms of HLH.
CASE PRESENTATION
Chief complaints
A 2-year-old boy was admitted to the hospital due to recurrent fever for > 20 d.
History of present illness
Approximately 20 d before admission, the patient presented with fever of 39.5°C without any inductive or provocative factors. Thrill, cough, vomiting and diarrhea were absent.
History of past illness
Persistent oral candidiasis and inoculation site infection were prominent in the past 2 years.
Personal and family history
No positive findings were obtained regarding his birth history and family history.
Physical examination
The patient was in acute distress with pale appearance. An ulcer was noted in the oral mucosa. No palpable lymphadenopathy was observed. His abdomen was distended,the liver descended about 6 cm below the costal margin and the spleen 5 cm below.Physiological reflexes were normal. The BCG vaccination scar was flushed and swollen (Figure 1A).
Laboratory examinations
A full blood count revealed pancytopenia with white blood cells of 1820/mm3,hemoglobin 58 g/L, and platelets 29000/mm3. C-reactive protein was 20.8 mg/L and procalcitonin was 1.53 ng/mL. Biochemical parameters indicated liver dysfunction with albumin 19.9 g/L, total bilirubin 73.5 mmol/L, triglyceride 3.26 mmol/L and ferritin 359.5 mg/L. Coagulation function test revealed that fibrinogen level was only 0.65 g/L. Blood culture was negative for bacteria. Serological studies were negative for Epstein–Barr virus (EBV), human immunodeficiency virus, TORCH syndrome, and parasite and antinuclear antibodies. Immunoglobulin and complement levels were normal. Sputum tuberculosis smear and T spot test forMycobacterium tuberculosiswere negative. Tests for fungal infection were positive with β-D-glucan 376.39 pg/mL and galactomannan 0.668. Bone marrow aspirate revealed a hypocellular image with an increased number of hemophagocytes. Serum T helper (Th) 1/Th2 cytokine determination demonstrated moderately elevated levels of interleukin (IL)-6 (43.8 pg/mL) and IL-10 (31.0 pg/mL) with normal IFN-γ concentration (2.6 pg/mL), which indicated probable infection but not HLH. Whole exome sequencing of peripheral blood revealed ade novoheterozygous variant, confirmed by Sanger sequencing, in the DNA binding domain of STAT1 (c.1154C>T) causing a T385M amino acid substitution in STAT1 protein, which was previously reported as a pathogenic GOF mutation(Figure 1B).
Imaging examinations
B-ultrasound showed hepatosplenomegaly. Chest computed tomography (CT)showed a cavity lesion in the lung (about 1.3 cm × 1.5 cm) (Figure 1C).
FINAL DIAGNOSIS
The final diagnosis was Mendelian susceptibility to mycobacterial disease (STAT1 defect) and HLH.
TREATMENT
The patient was firstly treated with ceftriaxone sodium followed by cefoperazone/sulbactam without efficacy. Voriconazole was administered after the β-D-glucan and galactomannan results were obtained. Cephalosporin antibiotics were replaced by meropenem. However, although the above intensive antimicrobial therapy was given febrile disease was not resolved. As this patient presented with prolonged fever,hepatosplenomegaly, pancytopenia, hypofibrinogenemia, hypertriglyceridemia and hemophagocytosis in the bone marrow, HLH was considered and dexamethasone and etoposide were administered according to the HLH-94 protocol on day 5 after admission. White blood cells and platelets gradually increased and hepatosplenomegaly was relieved but fever was still uncontrolled. Re-examination of chest CT revealed that the cavity in the lung was larger (2.2 cm × 1.5 cm) (Figure 1C).With informed consent, bronchofiberoscopy was performed andMycobacterium boviswas identified in bronchoalveolar lavage fluid by next-generation sequencing. The patient then received anti-tuberculosis therapy and the fever was resolved and all the disease-related signs disappeared.
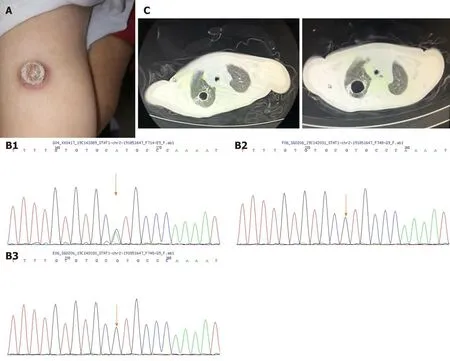
Figure 1 Clinical manifestation and results of gene sequencing. A: Bacille Calmette-Guerin vaccination scar was flushed and swollen; B: Sanger sequencing result of STAT1 of the patient (B1), father (B2) and mother (B3) indicated spontaneous mutation of STAT1(c.1154C>T); C: Cavitary lesion in the lung before and after antifungal therapy.
OUTCOME AND FOLLOW-UP
Hematopoietic stem cell transplantation was refused due to financial difficulty. During the following days, the patient had recurrent febrile diseases which were temporarily resolved by antibiotics or combined with a corticosteroid. He died six months later due to severe pneumonia.
DISCUSSION
The IFN-γ/IL-12 signaling pathway plays an important role in controlling infections with bacteria such as nontuberculous mycobacteria,M. tuberculosis, andSalmonella[4].Stimulation of IFN-γ receptors leads to phosphorylation of STAT1, which forms homodimers, translocates to the nucleus, and induces transcription of numerous genes[2]. Loss-of-function STAT1 mutations lead to severe bacterial, viral and mycobacterial infections, while dominant GOF mutations in STAT1 cause broad clinical phenotypes, including chronic mucocutaneous candidiasis (CMC, 98%),bacterial infections (74%), viral infections (38%), invasive fungal infections (10%),mycobacterial disease (6%) and autoimmune manifestations (37%)[1]. It is perplexing that mycobacterial disease occurs in patients with GOF STAT1 mutations, which was also seen in the present case. The underlying mechanism remains elusive, but impaired IL-17-producing T-cell development may partially account for this[5].Mutated STAT1 increases STAT1 phosphorylation by impairing nuclear dephosphorylation, and inhibits STAT3 and STAT4 functions and IL12R/IL-23R signaling, which leads to diminished Th1/Th17 and IL-12 responses and hence to increased susceptibility to various infections (Figure 2)[2,5,6]. Although GOF mutations enhance IFN-γ inducible target gene expression, re-stimulation with IFN-γ may give rise to impaired IFN-γ responses[7].
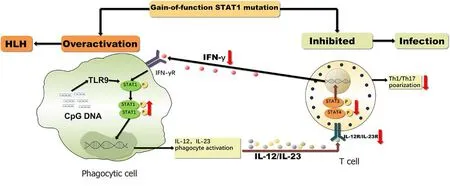
Figure 2 Pathophysiology of STAT1 gain-of-function mutation causing hemophagocytic lymphohistiocytosis and infection. In Mycobacterium bovis infection, Toll-like receptor 9 is activated by the pathogen, which then induces persistent phosphorylation of STAT1 due to its gain-of-function mutation, together with interferon-γ, leading to the overwhelming activation of phagocytic cells, resulting in hemophagocytic lymphohistiocytosis. In addition, increased STAT1 phosphorylation inhibits interleukin (IL) 12R/IL-23R signaling, leading to diminished T helper (Th) 1/Th17 and IL-12 responses, which then increase susceptibility to various infections. HLH: Hemophagocytic lymphohistiocytosis; TLR9: Toll-like receptor 9; IL-12: interleukin-12; Th1: T helper 1.
According to our previous studies, the inflammatory cytokine pattern of significantly elevated levels of IFN-γ and IL-10, with only modestly elevated IL-6 levels has high diagnostic accuracy for HLH[8]. However, the present patient only presented with slightly elevated IL-6 and IL-10 and IFN-γ level was within the normal range. Is IFN-γ secretion inhibited in patients with GOF STAT1 mutation? According to Smeekenset al[6], IL-12 induced no IFN-γ production in cells obtained from CMC patients, and IL-1β and IL-23 induced less IL-17 production, whereas production of tumor necrosis factor-α in response to IFN-γ and lipopolysaccharide was higher in peripheral blood mononuclear cells from CMC patients. This phenomenon is consistent with the normal IFN-γ level in our patient.
Another question to resolve is how HLH developed in a patient with normal IFN-γ.HLH is a severe hyperinflammatory syndrome induced by aberrantly activated macrophages and cytotoxic T cells, and IFN-γ is considered a cardinal cytokine in the development of HLH. Thus, it is interesting that HLH in this patient could be triggered independently of IFN-γ. The low IFN-γ level indicated that the cytotoxic T cells were not strongly activated due to impairment of IL12R/IL-23R signaling. Do macrophages drive the occurrence of HLH-like syndromes? Previous studies showed that HLH can also be induced by Toll-like receptor (TLR) 9 overstimulation[9].Mycobacteria are strong inducers of TLR2, 4 and 9, which can induce serine phosphorylation of STAT1[10]. Thus, in a patient with GOF STAT1 mutation andM.tuberculosisinfection, TLR9 is activated by the pathogen, which then induces persistent phosphorylation of STAT1, leading to the overwhelming activation of phagocytic cells,resulting in HLH (Figure 2).
Patients who present with GOF STAT1 mutation combined with HLH are rarely reported. Our search of PubMed revealed only three patients with GOF STAT1 who developed HLH[11,12]. The onset ages were 7-10 years, with STAT1 mutations of c.876C>A (p.D292E), c.1189 A>G (p.N397D) and c.208C>T (p.R70C). Two of the patients presented with combined immunodeficiency disease and all three patients showed classic presentation of HLH with increased inflammatory markers and ferritin.However, two of the former patients presented with refractory HLH and died of multiple organ failure after hematopoietic stem cell transplantation.
CONCLUSION
The clinical presentations of GOF STAT1 mutations are varied. CMC is common, and mycobacterial disease and HLH are occasionally found. These patients presenting with HLH show a different cytokine profile and treatment response to those with familiar HLH and EBV-related HLH. Next-generation sequencing is a helpful approach for identifying such patients.
 World Journal of Clinical Cases2020年23期
World Journal of Clinical Cases2020年23期
- World Journal of Clinical Cases的其它文章
- Understanding the immunopathogenesis of COVID-19: Its implication for therapeutic strategy
- What is the gut feeling telling us about physical activity in colorectal carcinogenesis?
- Latest developments in chronic intestinal pseudo-obstruction
- Correlation between ductus venosus spectrum and right ventricular diastolic function in isolated single-umbilical-artery foetus and normal foetus in third trimester
- Clinical efficacy of integral theory–guided laparoscopic integral pelvic floor/ligament repair in the treatment of internal rectal prolapse in females
- Treatment of Kümmell’s disease with sequential infusion of bone cement: A retrospective study