
AT1-AA通过氧化应激诱导心肌H9c2细胞自噬及凋亡
2020-04-03 09:34岳继萍张文婷张志楠靳文文焦向英
中国病理生理杂志 2020年3期
岳继萍, 王 瑾, 张文婷, 张志楠, 靳文文, 焦向英
(山西医科大学生理学教研室, 山西 太原 030001)
《中国心血管病报告2018》指出,未来十年我国心血管疾病患病人数仍加速增长,尽管针对目前已知原因所采取的治疗效果显著,但心血管病死亡率仍高占居民总死亡原因首位[1],提示还有一些未知因素参与心血管疾病的发生发展。近年来,人们在心血管类疾病如高血压性心脏病、冠心病和心衰等患者血清中均检测到血管紧张素Ⅱ 1型受体自身抗体(angiotensin Ⅱ type 1 receptor autoantibody,AT1-AA),且其AT1-AA阳性率和滴度都明显高于正常人[2-4]。有研究表明AT1-AA可以直接诱导心肌细胞凋亡[5],提示 AT1-AA 参与心血管疾病的发生发展。但是,其诱导心肌细胞凋亡的机制尚不清楚。
研究表明自噬(autophagy)与心脑血管等疾病关系密切。自噬是一种受调控的程序性死亡,是细胞内降解衰老蛋白、受损胞质的过程[6]。生理情况下自噬水平较低;机体应激时,自噬被激活,为机体提供能量并维持细胞稳态[7-8];而刺激持续存在时,过度激活的自噬会损伤细胞[9]。AT1-AA具有类血管紧张素Ⅱ(angiotensin Ⅱ,Ang Ⅱ)的激动样作用,Ang Ⅱ可以激活自噬,那么AT1-AA是否也可激活自噬?是否可以诱导心肌细胞凋亡?
研究表明,氧化应激可以导致细胞自噬及心功能异常[10-11],抑制氧化应激可以阻止细胞的进一步损伤。AT1-AA可以诱导氧化应激[12],而AT1-AA诱导的氧化应激与其诱导的自噬、凋亡之间是否有关有待验证。
材 料 和 方 法
1 材料
H9c2细胞购自中国医学科学院协和细胞库。DMEM培养基购自HyClone;胎牛血清(fetal bovine serum,FBS)购自Cell Max;0.25%胰酶购自索莱宝科技有限公司;血管紧张素Ⅱ1型受体(angiotensin Ⅱ type 1 receptor, AT1-R)抑制剂替米沙坦(telmisartan, TM)、自噬抑制剂3-甲基腺嘌呤(3-methyladenine,3-MA)和氧化应激抑制剂α-硫辛酸(α-lipoic acid,α-LA)购自Sigma;CCK-8检测试剂盒、蛋白酶抑制剂苯甲基磺酰氟(phenylmethanesulfonyl fluoride,PMSF)、凝胶配制试剂盒、辣根过氧化物酶标记的羊抗兔IgG和超敏ECL化学发光试剂盒均购自武汉博士德生物工程有限公司;谷胱甘肽过氧化物酶(glutathione peroxidase,GSH-Px)测定试剂盒、超氧化物歧化酶(superoxide dismutase,SOD)测定试剂盒和丙二醛(malondialdehyde,MDA)测试盒均购自南京建成公司;LC3抗体、Beclin1抗体、p62抗体、caspase-3抗体和β-actin抗体购自CST。
2 主要方法
2.1细胞培养与分组 将心肌H9c2细胞分为阴性对照(negative IgG)组、AT1-AA处理组、AT1-AA+3-MA处理组、AT1-AA+α-LA处理组和AT1-AA+TM处理组。
2.2AT1-AA的提纯 用生物合成的血管紧张素Ⅱ 1型受体胞外第二环抗原肽段AT1R-ECⅡ(序列为:165~191,IHRNVFFIENTNITVCAFHYESQNSTL)主动免疫SD大鼠8周,IgGs亲和纯化提纯AT1-AA,酶标仪检测其浓度。
2.3CCK-8法检测细胞活力 收集对数期细胞加入96孔板中,细胞贴壁后将培养基吸掉,换加含有不同梯度浓度的AT1-AA培养基分别作用12、24、36和48 h,避光加入10 μL CCK-8液,细胞孵育箱内继续孵育2 h,酶标仪450 nm处测定吸光度(A)值,按照公式[细胞相对活力(%)=(实验孔A值-空白孔A值)/(对照孔A值-空白孔A值)×100%]计算。
2.4Western blot检测蛋白水平 分别检测自噬相关蛋白LC3、Beclin1和p62的表达以及凋亡相关蛋白cleaved caspase-3/caspase-3的水平。收集细胞,加入80 μL RIPA裂解液和1 μL的PMSF吹散细胞,超声碎裂1 h,12 000 r/min 离心15 min,取上清。BCA法测细胞浓度做SDS-PAGE,转膜,封闭,孵育 I 抗和 Ⅱ 抗,ECL发光液进行曝光。曝光条带用ImageJ软件做灰度值分析,用β-actin作内参照。
2.5氧化应激指标的检测 收集对数期细胞,碎裂细胞,12 000 r/min离心15 min取上清。据氧化应激指标GSH-Px、SOD和MDA试剂盒说明书进行操作,检测相关数据。
3 统计学处理
用SPSS 24.0统计软件进行分析。数据均采用均数±标准误(mean±SEM)表示,多组均数间比较采用单因素方差分析(one-way ANOVA),组间两两比较采用SNK-q检验,以P<0.05为差异有统计学意义。
结 果
1 AT1-AA引起H9c2细胞活力下降,凋亡增加
与negative IgG组相比,随着AT1-AA浓度的升高,H9c2细胞的活力逐渐下降,在10-5mol/L时下降显著(P<0.01),见图1A;10-5mol/L的AT1-AA分别孵育H9c2细胞不同时长(12 h、24 h、36 h和48 h),与negative IgG相比,AT1-AA处理细胞24 h~48 h,细胞活力均显著下降(P<0.01),见图1A。以上结果提示AT1-AA可浓度和时间依赖性的降低心肌H9c2细胞的活力。10-5mol/L的AT1-AA处理H9c2细胞24 h,Western blot检测凋亡相关蛋白cleaved caspase-3/caspase-3的水平,结果显示,与negative IgG组相比,AT1-AA组H9c2细胞的cleaved caspase-3水平上升(P<0.01),见图1B,提示AT1-AA可以诱导心肌H9c2细胞凋亡。
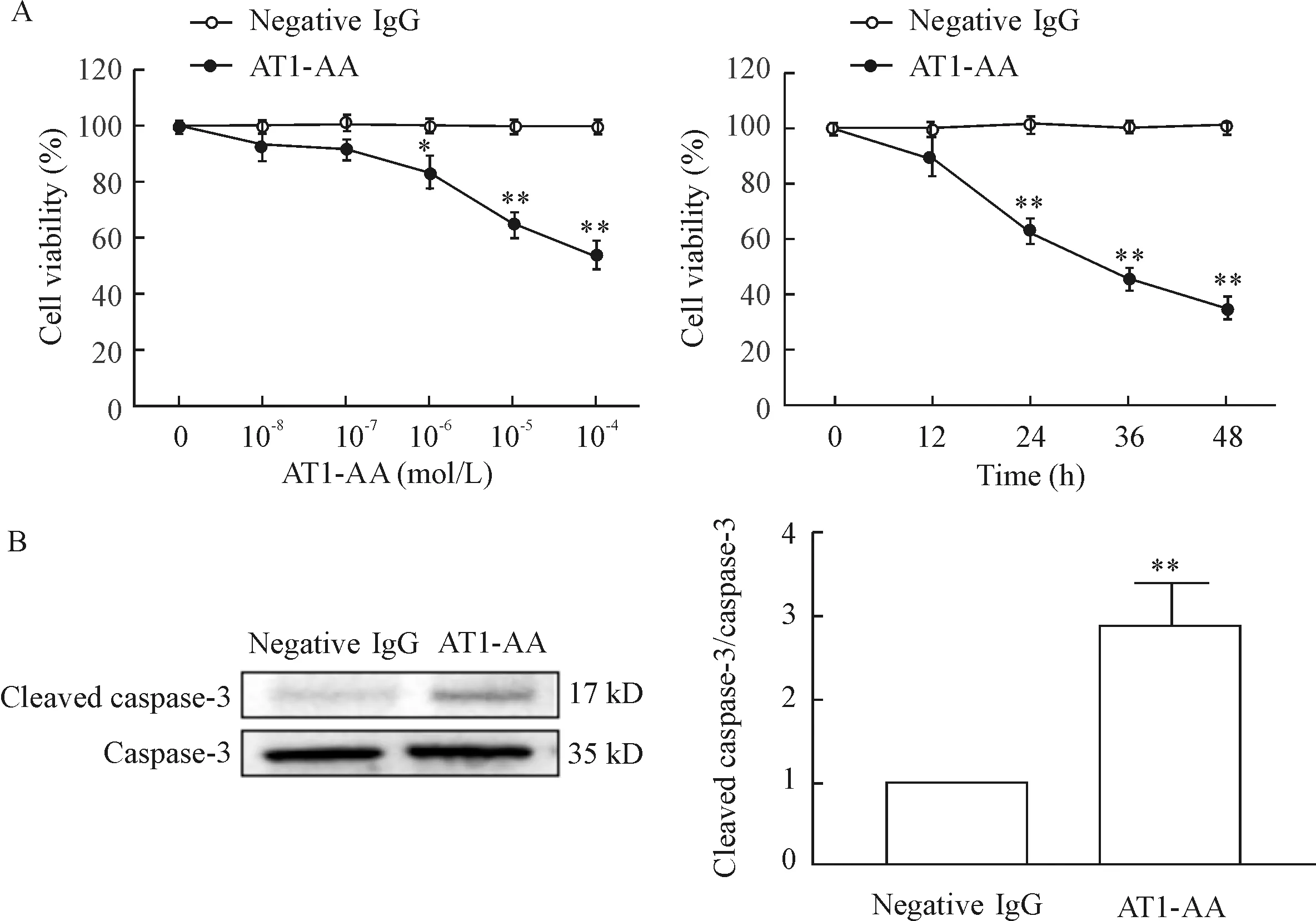
Figure 1.The changes of the viability (A) and the expression of cleaved caspase-3 (B) in the H9c2 cells induced by AT1-AA. Mean±SEM.n=6.*P<0.05,**P<0.01vsnegative IgG group.
图1 AT1-AA诱导的H9c2细胞的存活率及caspase-3的表达变化
2 AT1-AA诱导心肌H9c2细胞自噬增加
10-5mol/L AT1-AA处理细胞24 h后,Western blot检测自噬相关蛋白LC3、Beclin1和p62的蛋白水平,结果显示,与negative IgG组相比,AT1-AA组H9c2细胞的LC3-Ⅱ/LC3-Ⅰ和Beclin1蛋白的水平增加(P<0.01),而p62蛋白的水平则降低(P<0.01),见图2。这提示AT1-AA可以诱导心肌H9c2细胞自噬。
3 自噬抑制剂3-MA抑制AT1-AA诱导的H9c2细胞凋亡
10-5mol/L的3-MA预处理细胞1 h后,再加AT1-AA(10-5mol/L)处理细胞24 h。用Western blot检测自噬相关蛋白的表达水平,结果显示,与AT1-AA组相比,AT1-AA+3-MA组的H9c2细胞LC3-Ⅱ/LC3-Ⅰ和Beclin1蛋白的表达水平下降(P<0.05),而p62蛋白的表达上调(P<0.05),见图3A,提示AT1-AA诱导的自噬被抑制。Western blot检测凋亡相关蛋白cleaved caspase-3/caspase-3的水平,结果显示,与AT1-AA组相比,AT1-AA+3-MA组细胞的cleaved caspase-3/caspase-3水平下降(P<0.05),见图3B。以上结果提示抑制自噬可抑制AT1-AA诱导的H9c2细胞凋亡。
4 AT1-AA激活H9c2细胞的氧化应激
10-5mol/L的AT1-AA处理细胞24 h,收集细胞用氧化应激试剂盒检测GSH-Px、MDA和SOD的水平,发现AT1-AA组的GSH-Px和SOD高于negative IgG组,MDA低于negative IgG组(P<0.05),见表1。
5 氧化应激抑制剂α-LA降低AT1-AA诱导的H9c2细胞的自噬和凋亡水平
10-4mol/L α-LA预处理H9c2细胞12 h,再加AT1-AA(10-5mol/L)处理细胞24 h后收集细胞。氧化应激试剂盒检测GSH-Px、MDA和SOD的水平发现,与AT1-AA组相比,AT1-AA+α-LA组细胞的GSH-Px和SOD水平降低,而MDA水平升高(P<0.05),见表1。Western blot检测自噬相关蛋白LC3、Beclin1和p62的水平,结果显示,与AT1-AA组相比,AT1-AA+α-LA组H9c2细胞的LC3-Ⅱ/LC3-Ⅰ和Beclin1蛋白表达减少(P<0.05),而p62蛋白表达则上升(P<0.05),见图4A。Western blot检测凋亡相关蛋白cleaved caspase-3/caspase-3的水平,结果显示,与AT1-AA组相比,AT1-AA+α-LA组细胞的cleaved caspase-3水平下降(P<0.05),见图4B。以上结果提示氧化应激抑制剂α-LA可以抑制AT1-AA诱导的心肌H9c2细胞的自噬和凋亡。
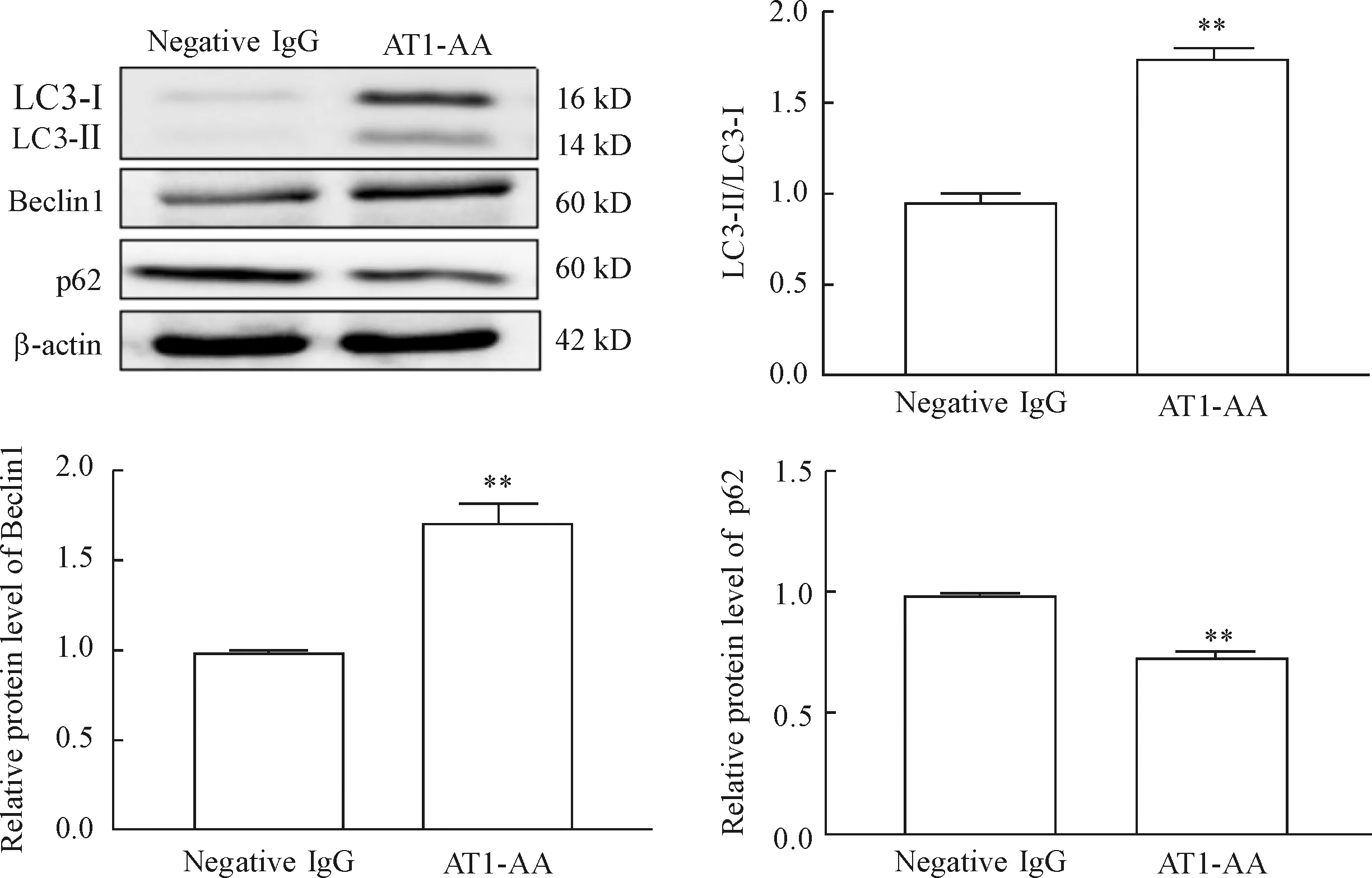
Figure 2.The autophagy of H9c2 cells was induced by AT1-AA. Mean±SEM.n=6.**P<0.01vsnegative IgG group.
图2 AT1-AA诱导H9c2心肌细胞的自噬增加
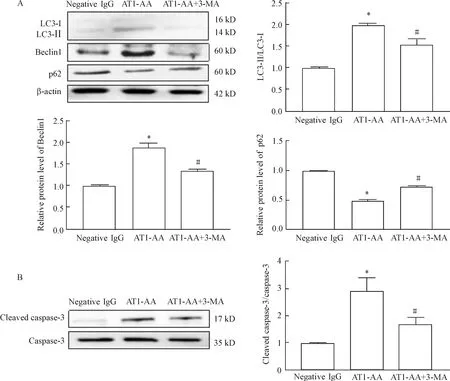
Figure 3.Autophagy inhibitor 3-MA reduced the protein levels of autophagy (A)- and apoptosis (B)- associated molecules in the H9c2 cells induced by AT1-AA. Mean±SEM.n=6.*P<0.05vsnegative IgG group;#P<0.05vsAT1-AA group.
图3 自噬抑制剂3-MA抑制AT1-AA诱导的H9c2细胞的自噬和凋亡的水平
表1 氧化应激水平的变化
Table 1.The levels of GSH-Px, MDA and SOD (Mean±SEM.n=10).
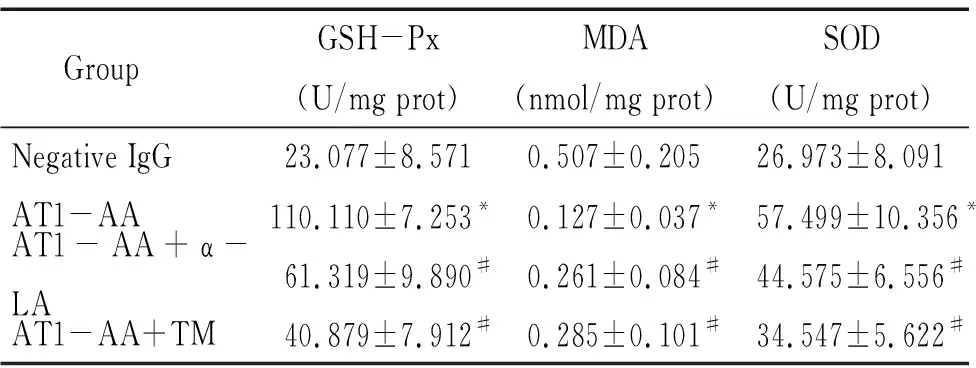
Group GSH-Px(U/mg prot)MDA(nmol/mg prot)SOD(U/mg prot)Negative IgG23.077±8.5710.507±0.20526.973±8.091 AT1-AA110.110±7.253*0.127±0.037*57.499±10.356*AT1-AA+α-LA61.319±9.890#0.261±0.084#44.575±6.556# AT1-AA+TM40.879±7.912#0.285±0.101#34.547±5.622#
*P<0.05vsnegative IgG group;#P<0.05vsAT1-AA group.
6 AT1-R抑制剂TM降低AT1-AA诱导的H9c2细胞氧化应激、自噬和凋亡水平
10-6mol/L的TM预处理细胞1 h,AT1-AA+TM再处理H9c2细胞24 h后收集细胞。氧化应激试剂盒分别检测GSH-Px、MDA和SOD的水平发现,与AT1-AA组相比,AT1-AA+TM组的GSH-Px和SOD水平降低,而MDA水平升高(P<0.05),见表1。Western blot检测LC3、Beclin1和p62的蛋白水平,结果显示与AT1-AA组相比,AT1-AA+TM组H9c2细胞的LC3-Ⅱ/LC3-Ⅰ和Beclin1蛋白表达减少(P<0.05),而p62蛋白表达则增加(P<0.05),见图5A。Western blot检测凋亡相关蛋白cleaved caspase-3/caspase-3的水平,结果显示,与AT1-AA组相比,AT1-AA+TM组细胞的cleaved caspase-3水平下降(P<0.05),见图5B。以上结果提示AT1-R抑制剂TM可以降低AT1-AA诱导的心肌H9c2细胞的氧化应激、自噬和凋亡水平。
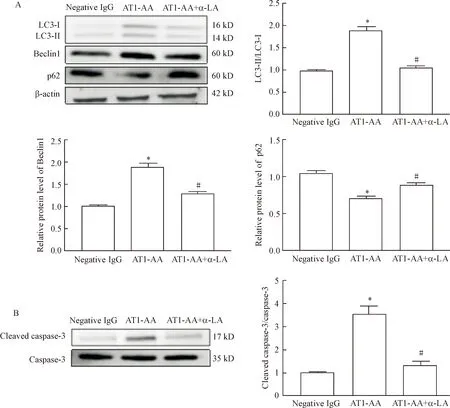
Figure 4.Oxidative stress inhibitor α-LA reduced the protein levels of autophagy (A)- and apoptosis (B)- associated molecules in the H9c2 cells induced by AT1-AA. Mean±SEM.n=6.*P<0.05vsnegative IgG group;#P<0.05vsAT1-AA group.
图4 氧化应激抑制剂α-LA降低AT1-AA诱导的H9c2细胞的自噬和凋亡水平
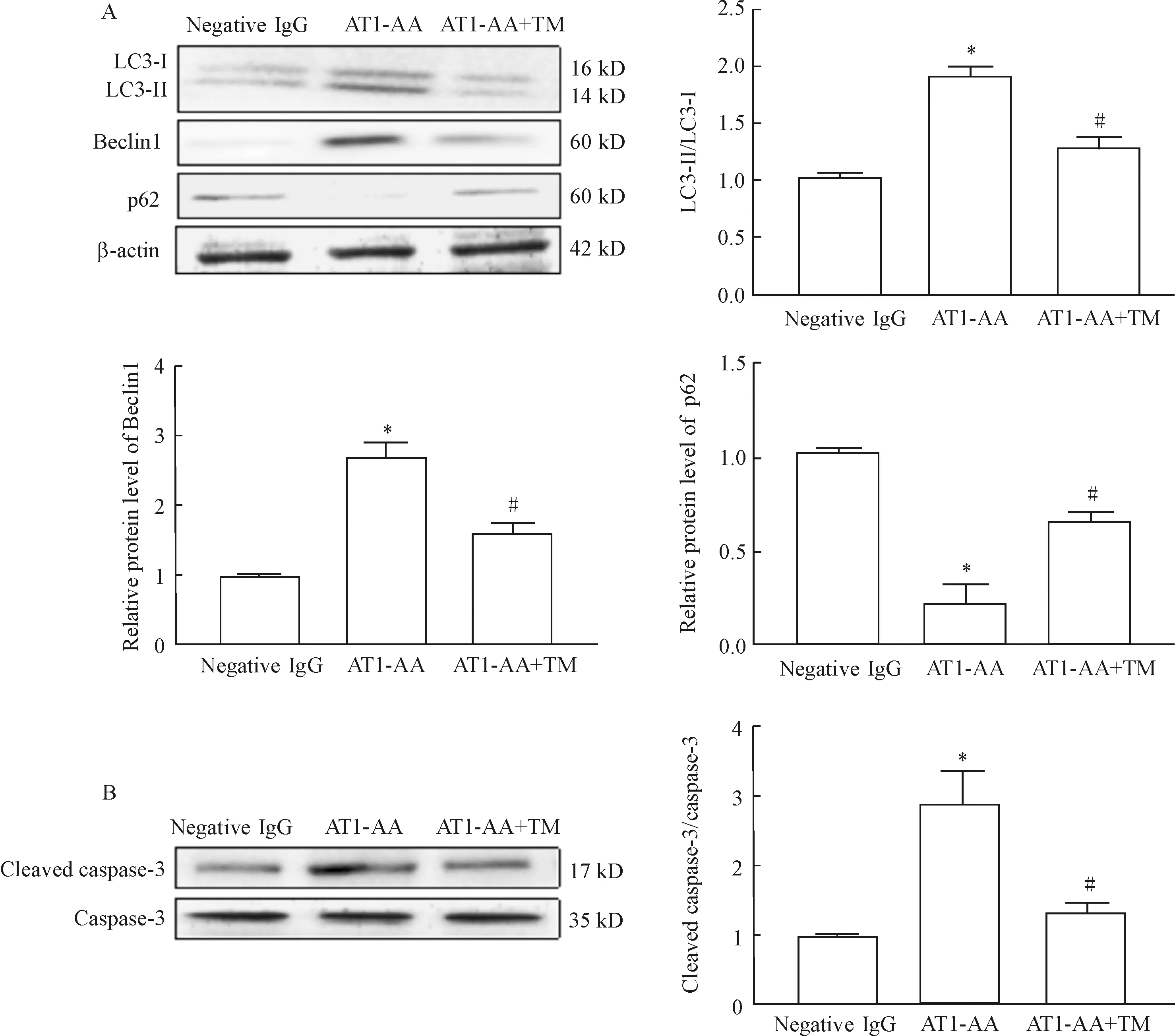
Figure 5.Telmisartan (TM) attenuated the protein levels of autophagy (A)- and apoptosis (B)- associated molecules in the H9c2 cells induced by AT1-AA. Mean±SEM.n=6.*P<0.05vsnegative IgG group;#P<0.05vsAT1-AA group.
图5 替米沙坦降低AT1-AA诱导的H9c2细胞的自噬和凋亡水平
讨 论
本研究首先用CCK-8法检测了AT1-AA可以浓度及时间依赖性降低H9c2细胞的活力;与negative IgG组相比,Western blot法检测显示AT1-AA组凋亡相关蛋白cleaved caspase-3/caspase-3的水平上升,提示AT1-AA可以导致心肌H9c2细胞活力下降,凋亡增加。而AT1-AA诱导细胞凋亡的机制不清。
自噬是溶酶体降解并清除受损细胞器的过程。通过平衡细胞合成和分解,自噬可以稳定细胞内环境。然而, 过度自噬可致细胞发生Ⅱ型程序性细胞死亡。自噬的程度可用LC3-Ⅱ/LC3-I比值评价,随着自噬发生,LC3-Ⅱ蛋白逐渐增加[13]。在自噬溶酶体降解过程中,p62受体作为底物结合物被降解清除,而当自噬受阻,p62蛋白的表达水平则显著上调[14]。Beclin1是哺乳动物参与自噬的特异性基因,可以调节自噬活性[15]。本研究通过用Western blot法检测自噬相关蛋白发现,AT1-AA促进LC3-Ⅱ/LC3-I和Beclin1的表达,而抑制p62蛋白的表达,表明AT1-AA可以激活自噬;为了进一步探讨自噬对AT1-AA诱导细胞凋亡的影响,我们用自噬抑制剂3-MA预处理细胞,发现AT1-AA对细胞的促凋亡作用被抑制。这说明自噬参与AT1-AA诱导H9c2细胞的凋亡过程。
氧化应激时细胞可通过自噬降解受损细胞器和长寿命蛋白质[16]。研究表明,自噬与氧化应激共同参与许多心脏疾病的发生发展,探讨自噬与氧化应激的可能关系有望成为心脏疾病的治疗新靶点[17-18]。研究表明AT1-AA可以激活氧化应激[19],与本研究的结果一致。而加入氧化应激抑制剂α-LA处理H9c2细胞发现,AT1-AA诱导的氧化应激被抑制,自噬和凋亡水平也有所下降,提示氧化应激位于自噬和凋亡的上游且调控AT1-AA诱导的自噬和凋亡过程。
研究表明,AT1-AA可以通过专一识别、结合AT1-R受体来发挥类血管紧张素Ⅱ的激动样作用且其对AT1-R的刺激作用不易脱敏[2, 20]。因此为了进一步验证AT1-R是否参与了AT1-AA诱导的氧化应激、自噬和凋亡,我们采用AT1-R抑制剂TM预处理细胞,结果表明AT1-AA诱导的氧化应激、自噬和凋亡水平明显降低,提示AT1-AA通过AT1-R影响H9c2细胞的氧化应激、自噬和凋亡。
综上所述,AT1-AA可以通过AT1-R激活氧化应激而上调自噬,进而导致心肌细胞的凋亡增加。
猜你喜欢
昆明医科大学学报(2022年4期)2022-05-23
上海交通大学学报(医学版)(2022年3期)2022-05-05
天津医科大学学报(2021年4期)2021-08-21
现代临床医学(2021年2期)2021-03-29
世界科学技术-中医药现代化(2020年2期)2020-07-25
中国报道(2018年2期)2018-04-20
安徽医科大学学报(2015年9期)2015-12-16
医学研究杂志(2015年12期)2015-06-10
癌变·畸变·突变(2015年3期)2015-02-27
