
慢性缺氧促心肌耐受急性缺血再灌注损伤的研究
2020-03-31 02:32:00
国际心血管病杂志 2020年2期
缺血性心脏病是常见的心血管疾病,其中紫绀型先天性心脏病患者长期处于严重乏氧状态[1-3],在手术中进行体外循环时提倡高浓度给氧,以保证氧供给,但高浓度补氧会加重心肌损害,不利于心肌功能恢复[4]。调节心肌血氧平衡、降低心肌负荷可缓解缺血再灌注后心肌损伤[5]。慢性缺氧处理是降低心肌缺血再灌注损伤的有效方法,心肌细胞会发生一系列慢性缺氧适应性变化,但具体机制尚未明确。本研究探讨慢性缺氧对心肌耐受缺血再灌注损伤的影响,以期为缺血性心脏病的防治提供依据。
1 材料与方法
1.1 实验动物、设备及试剂
60只体质量为140~180 g的雄性SD大鼠由成都医学院实验动物中心提供。高频彩色超声仪(Vevo 2100)、呼吸机(DH-150)购自加拿大VisualSonic公司;Langendorff灌流系统、多功能生理记录仪、全自动生化分析仪购自美国BIOPAC公司。戊巴比妥钠、肝素钙购自江苏常州千红生化制药股份有限公司,乳酸脱氢酶(LDH)试剂盒、肌酸激酶同工酶(CK-MB)试剂盒购自重庆川东化工有限公司化学试剂厂。
1.2 动物分组
将60只SD大鼠随机等分为实验组和对照组,每组30只;再根据在体实验及离体实验分别将实验组、对照组随机等分,分为在体实验组、在体对照组、离体实验组和离体对照组,每组15只。4组大鼠均给予同质饲料及饮用水,喂养28 d。实验组大鼠在模拟海拔5 000 m的动物低压低氧舱(成都医学院第一附属医院提供)中生活,舱内条件:大气压54 kPa,氧分压11.33 kPa,温度25 ℃。动物在舱内自由摄食饮水,每3~4 d将舱内气压调整1次,至相当于海拔3 000 m,时间1 h。对照组大鼠在正常氧环境中喂养。
1.3 构建在体缺血再灌注损伤模型
予以大鼠面罩吸氧,连接呼吸机,频率50次/min,潮气量2 mL/kg,吸呼比1∶2。备皮后,经左侧第3肋间隙进入并撑开胸腔,仔细暴露心脏并辨别出左冠状动脉前降支,将带针缝线从前降支下穿过,在血管上方放置直径1~2 mm聚乙烯管后打结。结扎后远端血管支配的区域颜色变苍白,表明成功结扎血管并致心肌组织缺血。0.5 h后剪断缝线,撤去聚乙烯管,观察心脏缺血区域颜色恢复为粉红色,关闭胸腔、复苏大鼠。
造模24 h后行超声心动图检测,随后立即开腹,通过下腔静脉注射100 IU肝素钙并采血5 mL以检测心肌酶,迅速摘除心脏,拭净血液后称重。
1.4 超声心动图检测在体大鼠心功能
造模前24 h及造模后24 h,腹腔注射戊巴比妥钠(2 mL/kg)麻醉大鼠,对大鼠称重并固定于动物手术台上,采用高频彩色超声仪13~24 MHz超声探头(MS250)检测大鼠心率(HR)、舒张期室间隔厚度(IVSd) 、收缩期室间隔厚度(IVSs)、舒张期左室后壁厚度(LVPWd)、收缩期左室后壁厚度(LVPWs)、左室射血分数(LVEF)、左室缩短分数(LVFS)、左室舒张末期容积(LVEDV)、左室收缩末期容积(LVESV),计算出每搏输出量(SV),SV=LVEDV-LVESV。
1.5 构建离体心脏再灌注损伤模型
连接Langendorff灌流系统和多功能生理记录仪,将压力换能器与球囊相连接。向K-H溶液中通入95%O2、5%CO2混合气体,使K-H溶液pH值维持在7.4左右,用K-H溶液冲洗整个Langendorff灌流系统。称重SD大鼠,3%戊巴比妥钠(2 mL/kg)腹腔注射麻醉后开腹,经下腔静脉注射100 IU肝素钙后取出心脏,在4 ℃ K-H溶液中洗净残余血液,迅速挂在Langendorff 灌流系统中,以80 cmH2O压力行主动脉逆行灌注。待心脏复跳后,将压力换能器和球囊经左心耳放入左心室,缓缓向球囊内注水,使左室舒张末期压力维持在4~10 mmHg,记录心室内压力变化。待心跳稳定、心率>200次/min并持续20 min后,记录心脏灌流液流出量,取500 μL灌流液置于EP管中,于-80 ℃冰箱保存,停止心脏灌注。30 min后打开灌流系统进行再灌注,每10 min记录一次心室内压力,60 min时取500 μL灌流液于EP管中,于-80 ℃冰箱保存待测,并摘除心脏。
1.6 心肌TTC染色和心肌梗死面积评估
TTC染色步骤:对心室进行切片,厚度约为0.5 cm,将心肌片放入1% TTC溶液中,37 ℃恒温水浴加热20 min、10%多聚甲醛固定30 min后取出照相。采用ImageJ软件测量,心肌梗死面积=心肌片梗死面积/心室肌总面积。
1.7 LDH和CK-MB活性检测
将血液标本及灌流液标本3 000转/min离心10 min,取上清液作为检测样品。采用全自动生化分析仪检测LDH和CK-MB活性,操作严格按照试剂盒检测说明书进行。
1.8 统计学分析
采用SPSS 17.0软件进行统计学分析,所有数据以均数±标准差表示,经 Kolmogorov-SmirnovD检验分析均为正态分布,组间比较采用独立样本t检验,组内比较采用配对样本t检验。P<0.05为差异有统计学意义。
2 结果
2.1 慢性缺氧对在体心脏缺血再灌注损伤的影响
2.1.1 在体实验组与在体对照组大鼠缺血再灌注损伤前后超声心动图指标比较 缺血再灌注前,在体实验组HR、LVESV均明显大于在体对照组(P均<0.05),IVSd、IVSs、LVPWd、LVPWs、LVEF、LVFS、LVEDV、SV均明显小于在体对照组(P均<0.05)。在体实验组和在体对照组经历缺血再灌注后大鼠的心功能均有不同程度损伤。在体实验组缺血再灌注后HR、IVSd、IVSs、LVPWd、LVPWs、LVEDV、LVESV均有不同程度上升(P均<0.05),而LVEF、LVFS、SV有不同程度下降(P均<0.05);在体对照组缺血再灌注处理后HR、LVEDV、LVESV有不同程度上升,IVSs、LVPWd、LVPWs、LVEF、LVFS、SV较缺血再灌注前明显下降(P均<0.05)。 缺血再灌注后,在体实验组HR、IVSs、LVPWd、LVPWs、LVEF、LVFS均明显大于在体对照组(P均<0.05),IVSd、LVEDV、LVESV、SV均明显小于在体对照组(P均<0.05)。见表1。
2.1.2 在体实验组与在体对照组大鼠缺血再灌注损伤后心肌梗死面积及血清心肌酶水平比较 TTC染色后可见实验组大鼠心肌梗死面积较对照组明显减少[(0.07±0.02)%对(0.16±0.01)%,P<0.05],见图1。与对照组相比,实验组CK-MB活性明显降低[(176.14±20.82)IU/L对(658.83±74.18)IU/L,P<0.05];两组LDH活性无统计学差异[(610.86±72.20)IU/L对(574.67±63.35)IU/L]。
2.2 慢性缺氧对离体心脏缺血再灌注损伤的影响
2.2.1 离体实验组与离体对照组大鼠离体心脏缺血再灌注损伤前后心室压力指标比较 压力换能器测定缺血再灌注损伤前后心室内压力变化指标大鼠左室发展压(LVDP)、左室舒张末期压力(LVEDP)变化情况,以及最大收缩速率[max(dp/dt)]、最大舒张速率[min(dp/dt)]的变化。缺血再灌注损伤前,离体实验组LVDP低于离体对照组(P均<0.05),两组LVEDP的差异无统计学意义;经历缺血再灌注损伤,离体实验组及离体对照组LVDP均较缺血再灌注前下降,LVEDP均较缺血再灌注前上升(P均<0.05);缺血再灌注损伤后,离体实验组LVDP明显高于离体对照组,LVEDP明显低于离体对照组,(P均<0.05)。缺血再灌注损伤前,离体实验组反映心室内压力变化快慢的指标max(dp/dt)、min(dp/dt)均明显低于离体对照组(P均<0.05);经历缺血再灌注损伤后离体实验组及离体对照组max(dp/dt)、min(dp/dt)均有不同程度下降,差异有统计学意义(P均<0.05);缺血再灌注损伤后,离体实验组max(dp/dt)、min(dp/dt)均明显高于离体对照组(P均<0.05)。见表2。

表1 两组大鼠超声心动图指标比较
注:与在体对照组缺血再灌注前比较,(1)P<0.05;与在体对照组缺血再灌注后比较,(2)P<0.05;与在体实验组缺血再灌注前比较,(3)P<0.05
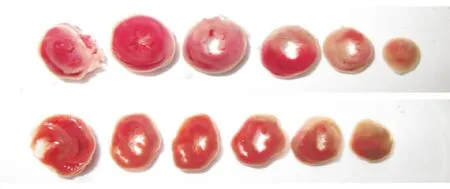
注:灰白色区域为心肌梗死区,上图为在体对照组,下图为在体实验组

表2 缺血再灌注前后两组大鼠离体心室压力指标比较
注:与离体对照组缺血再灌注前比较,(1)P<0.05;与离体实验组缺血再灌注前比较,(2)P<0.05;与离体对照组缺血再灌注后比较,(3)P<0.05
2.2.2 离体实验组与离体对照组组大鼠离体心脏缺血再灌注损伤后心肌梗死面积、LDH活性比较 TTC染色可见实验组心肌梗死面积较对照组明显减少[(0.08±0.01)%对(0.13±0.02)%,P<0.05];实验组LDH活性较对照组明显降低[(4.00±0.37)IU/L对(7.33±0.99)IU/L,P<0.05]。
3 讨论
心肌慢性缺氧是紫绀型先天性心脏病、肺源性心脏病、冠状动脉粥样硬化性心脏病、心肌肥厚等疾病的共同病理生理过程,研究慢性缺氧心肌的适应性反应及机制,对上述疾病的治疗具有重要意义。
慢性缺氧不仅可以使心肌细胞超纤维结构发生变化,导致心肌重构及心脏收缩功能下降,还可以促进血管活性物质分泌,血管内皮细胞增殖,使肺动脉压力升高,导致右心衰竭,引起慢性缺氧相关性心肌病。心肌细胞为终末分化细胞,缺氧会导致心肌细胞凋亡增加,但心肌细胞有较强的自我适应及修复能力,可在应激状态下促进心肌细胞存活,维持心肌细胞数量,保存心脏功能。1986年,Murry等[6]首次提出慢性缺氧适应是心肌一种重要的自我适应能力,缺氧预处理可预防缺血和缺血再灌注损伤等对心肌造成的伤害。慢性缺氧时心肌细胞产生一系列结构、功能、代谢变化,能增强心肌对急性缺氧导致的心肌梗死、收缩功能不良、室性心律失常等的耐受力[7-8]。研究发现,慢性缺氧可增加血管内皮生长因子的表达及线粒体数量,提高心肌组织供血及能量合成,使心肌能最大程度利用有限氧[9-10]。肖颖彬等[11]研究发现,慢性缺氧时,心肌细胞可调节代谢相关激酶的活性,增加葡萄糖利用并减少脂肪酸利用;Peng等[12]研究发现,慢性缺氧大鼠心肌细胞可以通过上调抗凋亡蛋白Bcl-2的表达,降低促凋亡蛋白Bax的表达水平,抑制缺血再灌注诱导的心肌细胞凋亡。通过以上多种适应机制,慢性缺氧增强心肌细胞耐受应激的能力。
再灌注虽可以增加缺血心肌的血流,挽救心肌,但同时又可造成不可逆转的附加损伤[13]。研究表明,长时间的缺血再灌注损伤会导致心肌细胞死亡及功能障碍[14],探究缺血再灌注损伤机制,对减少心肌损伤、降低缺血性心脏病的发生有重要意义。再灌注损伤的具体机制尚未明确,很多学者认为其主要通过氧化应激、线粒体功能异常导致心肌能量代谢异常和炎性反应,从而造成心肌细胞功能障碍[15]。研究表明,缺血再灌注损伤时氧自由基增多[16],心肌细胞的生物膜结构受到破坏,蛋白质功能受到抑制,其核酸和染色体也受到损害[17],最终导致心肌细胞功能改变,引起心肌损伤。同时,缺血再灌注损伤可使线粒体功能受损,细胞色素氧化酶活性受抑,氧自由基生成增加[18]。缺血时心肌细胞三磷酸腺苷(ATP)迅速减少,再灌注时由于氧自由基的损伤,阻碍了心肌细胞氧化磷酸化的过程[19],双重作用下心肌耐受能力降低,从而造成细胞损伤。
临床上缺血再灌注损伤比较常见,冠状动脉粥样硬化性心脏病患者经皮冠状动脉介入术、非体外循环下冠状动脉旁路移植术等均发生了在体缺血再灌注损伤的病理生理过程。常规体外循环下心脏手术停跳复跳过程是典型的缺氧-复氧过程,相当于离体缺血再灌注损伤的病理生理过程。因此,本研究分别制作了大鼠在体、离体缺血再灌注损伤模型,研究发现,慢性缺氧预处理能使缺血再灌注损伤后心肌梗死面积减少,心功能改善,从而减少心肌细胞在缺血再灌注后的损伤,维持心肌细胞更高的存活率。研究表明,再灌注时氧分压越高,产生的氧自由基就越多,对心肌的损伤也越大;低氧分压能使再灌注末的缺血心肌产生较少的氧自由基,显著提高细胞抗氧化酶活性,使线粒体等超微结构的损害减轻[20]。因此,我们认为低氧减轻缺血再灌后心肌的损伤是由于低氧状态下氧自由基生成减少,但其中涉及的具体信号通路尚未明确,需进一步研究。
猜你喜欢
中国典型病例大全(2022年11期)2022-05-13 15:12:17
中华养生保健(2020年7期)2020-11-16 01:14:24
天津医科大学学报(2019年6期)2019-08-13 07:04:34
山东林业科技(2019年2期)2019-06-03 10:10:54
现代园艺(2017年11期)2017-06-28 11:32:46
奥秘(2016年10期)2016-12-17 13:13:11
实用临床医学(2016年8期)2016-06-07 01:28:18
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:38
西藏科技(2015年9期)2015-09-26 12:15:31
中国当代医药(2015年10期)2015-03-01 02:02:30
