
基于全基因组重测序技术鉴定转基因大豆插入位点的方法*
2020-03-31 02:31:54郭兵福郭勇洪慧龙邱丽娟
大豆科技 2020年1期
郭兵福,郭勇,洪慧龙,,邱丽娟**
(1.中国农业科学院作物科学研究所/国家农作物基因资源与遗传改良重大科学工程,北京 100081;2.东北农业大学农学院,哈尔滨 150030)
明确的插入位点及其旁侧序列是转基因材料进入生物安全评价更高阶段的必要条件。传统的插入位点鉴定方法以基于已知外源序列的PCR扩增技术为基础,主要有TAIL-PCR和Genome walking等。大豆是一个古四倍体农作物,近75%的基因以多拷贝或复杂拷贝的形式存在,因此,传统的以PCR为基础的插入位点鉴定方法在转基因大豆中工作效率并不高。以自主研发的转G2EPSPS和GAT基因抗草甘膦转化体ZH10-6和GE-J16为研究对象进行重测序,数据过滤后成对的Pariedend Reads分别拆分为单个独立的Read,每个Read均单独与参考基因组和载体序列进行比对,将一端比对至载体上,一端比对至参考基因组上的复合序列(Junction reads)进行富集,根据富集结果预测转基因大豆的插入位点,并利用PCR扩增测序和遗传共分离分析方法对预测的插入位点进行验证(图1)。结果发现,外源T-DNA插入在转化体GE-J16的19号染色体50543767-50543792位和ZH10-6的17号染色体7980527-7982541位间,TDNA的整合分别导致了基因组上24 bp和13 bp碱基序列的删除(图2)。建立了利用重测序技术鉴定大豆插入位点的方法,与前人研究进展相比,对序列比对方式进行了优化,由前人研究的Pairedend Reads成对比对调整为单个Read独立比对的方式,在测序深度大幅降低的条件下亦能精确鉴定转基因大豆的插入位点及其旁侧序列,为利用重测序技术经济便捷高通量鉴定转基因大豆插入位点提供参考。

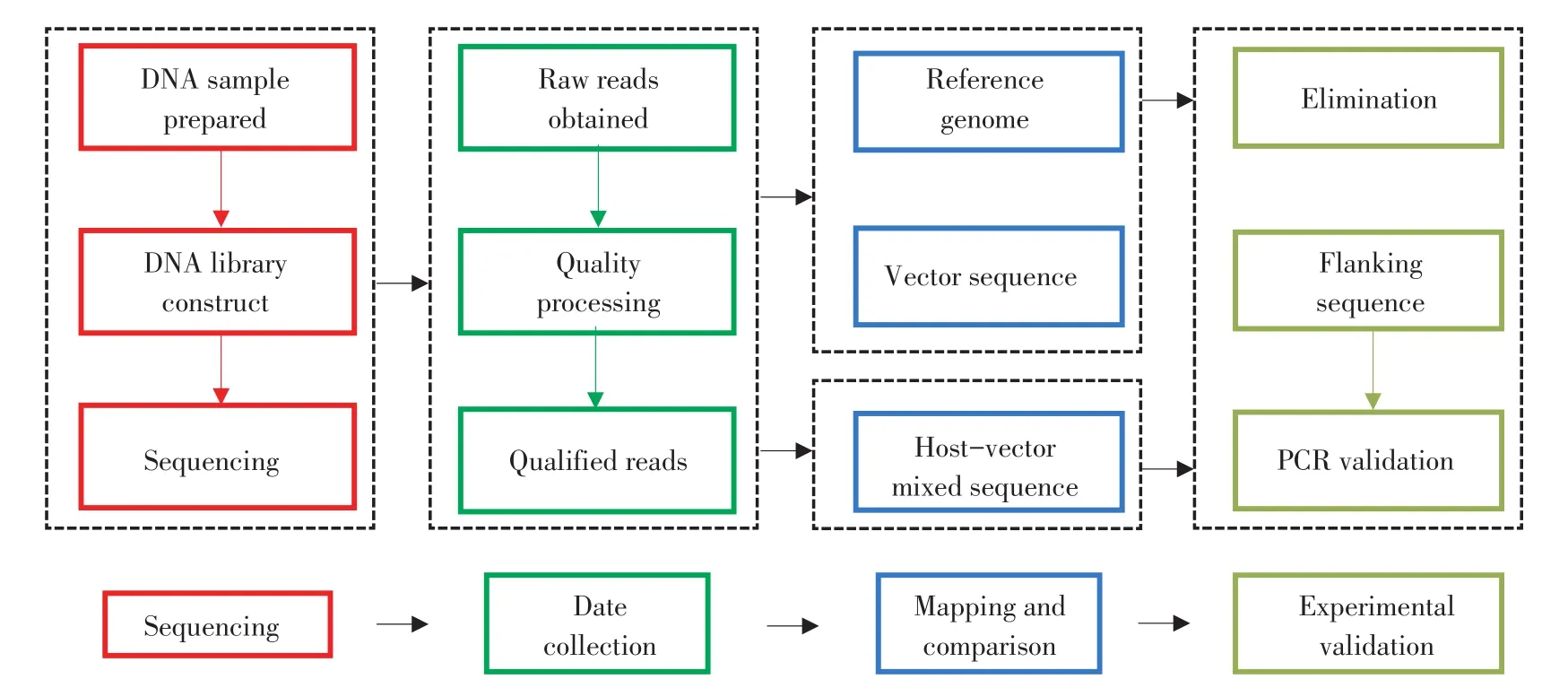
图1 利用重测序技术鉴定大豆插入位点技术流程
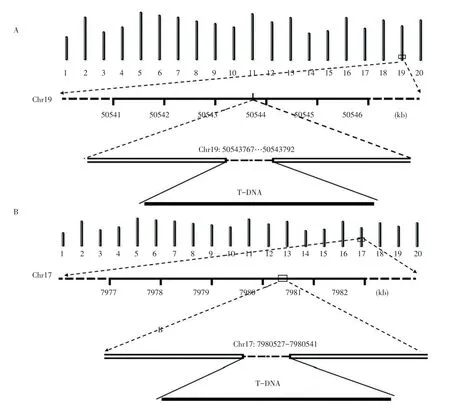
图2 转基因大豆GE-J16(A)和ZH10-6(B)精确插入位点示意图
(Front.Plant Sci,2016,doi:10.3389/fpls.2016.01009)
猜你喜欢
学与玩(2022年10期)2022-11-23 08:32:00
今日农业(2022年3期)2022-06-05 07:12:08
今日农业(2021年11期)2021-08-13 08:53:24
中成药(2018年10期)2018-10-26 03:41:34
中国继续医学教育(2015年1期)2016-01-06 01:36:01
创新科技(2015年1期)2015-12-24 06:23:21
治淮(2015年6期)2015-12-24 06:07:52
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
江苏卫生事业管理(2014年2期)2014-02-28 01:59:54
