
髓母细胞瘤的分期和分型的研究进展
2020-03-27 07:45:30陈立华孙恺陈文锦夏勇张洪钿徐如祥
中华脑科疾病与康复杂志(电子版) 2020年5期
陈立华 孙恺 陈文锦 夏勇 张洪钿 徐如祥
髓母细胞瘤(medulleblastoma,MDB)是Cushing和Bailey于1925年首先提出,是发生在小脑蚓部的小细胞分化不良的恶性肿瘤[世界卫生组织(World Health Organization,WHO)Ⅳ级][1]。多数学者认为MDB起源于小脑蚓部或小脑、第四脑室底部的外颗粒层细胞,亦有认为MDB起源于室管膜下基质细胞[2]。约30%~60%的MDB患者有等臂染色体17q的异常及1号染色体异常,17p、18p和11p最常发生缺失,而7q、i17q、18q、7p、13q和18p常见增加,17p的丢失,提示预后极差[3]。MDB整体预后良好,但取决于组织病理学类型、分期和分子分型。
一、组织病理
WHO将MDB定义为一种恶性的、侵袭性的小脑胚胎性肿瘤,起源于小脑或后颅窝(来自第四脑室或蚓部),具有神经元分化和通过脑脊液途径扩散转移到整个大脑和脊柱的固有趋势。
(一)镜下特征
肉眼观察,MDB呈灰红色或粉红色、质地软而脆,一般边界较清楚,肿瘤无包膜,一般没有中央坏死和出血,少数有小囊变和散在钙化。镜下表现为细胞密集,细胞核深染,胞浆稀少。镜下观察,典型的MDB呈淡蓝色,胞浆呈深蓝色,细胞大多未分化。不到50%的MDB可见Homer-Wright花环图案,由一个圆形的核和细胞质组成。Ki-67/MIB1抗体阳性染色可检测到高达80%的肿瘤有丝分裂。成人MDB患者中约50%为促结缔组织增生型MDB,而儿童只有15%。
(二)组织学分型
2007年和2016年WHO根据组织病理学上的差异性,将MDB分为4个组织学类型:经典型MDB(classic MDB,CMB)、大细胞/间变型(large cell/anaplastic,LC/A)MDB、弥散结节型或广泛结节型MDB(MDB with extensive nodularity,MBEN)、促纤维增生/结节型MDB(desmoplastic/nodular variant of MDB,DNMB)。这些亚型的组织病理特点和预后各不相同(表1),MBEN的预后优于LC/A肿瘤。
1.CMB:细胞密集排列,细胞核呈圆形、椭圆形和胡萝卜形,且浓染。经典型MDB的5年总生存率(overall survival,OS)可达78%。
2.DNMB:多见于成人小脑半球。儿童DNMB型预后相对较好,存活率很高。<3岁的MDB患儿中,DNMB发生率约为44%(32%~61%);3~5岁时发病率下降到10%~20%。
3.MBEN:多发生于Rutkowski等[4]报道的一项荟萃分析中,21例MBEN患儿的8年无事件生存率和OS分别为86%和95%,87例DNMB患儿分别为48%和72%。该型婴幼儿神经元分化明显、侵袭性不高,预后较好,比CMB结局更好,5年OS可达82%。
4.LC/A:有大、圆形或多形态的核,核仁突出,胞浆丰富。LC/A具有侵袭性生物学特性,对治疗反应不佳,容易复发或转移,5年OS只有44%,传统上认为LC/A是预后最差的组织学类型,与MYC基因过度表达有关。

表1 髓母细胞瘤的组织学分型
传统上根据MDB的病理类型来制定综合治疗方案、预测肿瘤复发和转移的风险。DNMB、CMB的患者预后较好,而LC/A常常有脑脊液播散,尽管较少见,但预后差。有时同为高风险组的MDB,也接受相同或相似治疗,可能有不同的临床转归,说明仅仅依靠病理分型来制定治疗方案和预测预后是不精确的。
(三)分期
MDB分期标准包括术后72 h MRI复查来评估肿瘤切除的程度、术后2周脊髓MRI和腰穿脑脊液细胞学检查来评估肿瘤是否发生种植和转移。必须同时进行脊髓增强MRI和腰穿脑脊液细胞学检查,缺一项检查有可能遗漏约15%的脑脊液播散病例。1969年Chang等[5]曾报道的手术分期系统仍然是目前常用的风险分层标准,以此预测治疗结果并指导制定治疗计划。M0期,无蛛网膜下腔肿瘤转移证据;M1期,脑脊液细胞学检查阳性、神经轴影像学上没有肉眼可见的软脑膜肿瘤沉积;M2期、M3期、M4期,颅内、脊髓或颅外发现结节性转移灶。M分期的界定主要依赖于神经影像学检查和脑脊液细胞学检查,术前完成评估是最理想的,术后神经影像和脑脊液细胞学检查会受到手术的影响。术后残留的体积、患者的年龄、病理变异和M分期是预后的良好预测因素,可以对患者进行风险分层。高危患者具有以下特征之一:年龄≤3岁,最大安全切除后残留肿瘤≥1.5 cm2,LC/A型MDB,M1~M4分期;没有高风险组任何特征者为低风险组。
二、分子分型
根据病理组织学和临床放射学风险分级制定治疗方案,有导致无法预测的复发和治疗失败的风险。细胞遗传研究发现MDB有不同的分子亚群,根据激活的信号通路不同,2016年的中枢神经系统WHO分类中将MDB分为4个不同的亚群:WNT激活型、SHH型(SHH激活和TP53突变型,SHH激活和TP53野生型)、非WNT/非SHH第3、4组,这些亚型有显著异质性,具有不同的基因组学、临床特征和生物学特性[6]。需要根据MDB的分子风险分层,为每例患儿制定个性化分层治疗方案具有重要的意义,以期提高生存率和减少复发[6]。同一病理类型的MDB有不同的遗传背景,预后也可能有所不同,MDB的分子分型有可能推动新的靶向治疗。
(一)分子分型的特征
MDB具有高度异质性,组织病理分类并不能很好地阐述预后的差异性,DNA水平变异可能才是MDB发生的原因。核内β-catenin、TrkC的mRNA表达水平、MYC扩增水平及染色体等可预测MDB患者的治疗转归。新的分子分型影响MDB现有的诊断标准和治疗方案。不同分子亚型的MDB具有不同的临床特征和病理组织学特征,以及不同的临床预后。这些亚组已明确识别出其有显著的特征差异(表2)[6]。对这些亚组的深入研究,使得基于这些肿瘤分子亚型的风险分层成为可能,并为制定MDB的治疗方案提供新的依据。
(二)肿瘤的发生机制
YAP1是癌基因转录的辅助启动子,能促进细胞增殖和转化,在MDB的WNT和SHH亚型中高表达,而在非WNT/非SHH亚型MDB中[7]。GAB1在SHH型信号通路中具有特异性。YAP1蛋白作为WNT和SHH MDB亚型的特异性标志物,而GAB1作为SHH MDB亚型的特异性标志物。WNT MDB除Yap1免疫反应性外,还显示肿瘤细胞核内β-catenin蛋白的积聚,并表达Otx2[8]。因此,可以通过一组免疫组化标志物安全地进行鉴别。
1.WNT型:最常见的遗传改变是6号染色体单体、编码β-catenin蛋白的CTNNBl基因稳定突变、DDX3X和TP53基因突变[9]。该亚型肿瘤均存在明显的WNT信号通路的异常激活,致使该通路效应因子β-catenin在肿瘤细胞中大量积累并逐步进入细胞核内表达,然后通过与T细胞因子/淋巴增强因子的相互作用,进一步激活下游靶基因,从而引起该亚型肿瘤的发生[10]。WNT信号通路的异常激活导致WIF、DKK、DKK2等相关抑制因子发生负反馈调节[10]。在85%的儿童WNT型MDB中,6号染色体存在1个拷贝的缺失,即6号染色体单体,这是该亚型的特征性标志[3]。
2.SHH型:SHH激活MDB可细分为TP53突变型和TP53野生型,表达p75NGFR和Gab1等特异性靶蛋白,与WNT MDB共同表达核Yap1,但缺乏Otx2表达。SHH型的发生与编码SHH通路上游肿瘤抑制蛋白PTCH1的基因突变密切相关。PTCH1的缺失可导致SHH通路异常激活,进而促使MDB的形成。染色体9q缺失和MYCN、YAP1的扩增是SHH亚型的标志。SHH型突变发生率最高,由于对治疗决策和可能的生殖系改变有重要影响,应在认证的实验室中用有效的方法对所有SHH激活的MDB进行TP53测序,以明确区分有无TP53改变的SHH激活MDB。TP53突变型SHH MDB通常表现为LC/A,可能与MYCN和GLI2扩增及17p缺失有关;TP53野生型SHH MDB更常见于DNMB型,可能与PTCH1缺失和10q缺失有关。根据SHH亚型生物学特征的差异,进一步细分为SHHα、SHHβ、SHHγ、SHHδ 亚群。SHHα亚型主要发生于3~17岁的儿童,其特征是TP53突变联合MYCN扩增,该型极易发生转移,预后最差;SHHδ 亚型主要见于成人,预后相对较好。Tabori等[11]发现p53累积对预测MDB的TP53突变的敏感性为100%。
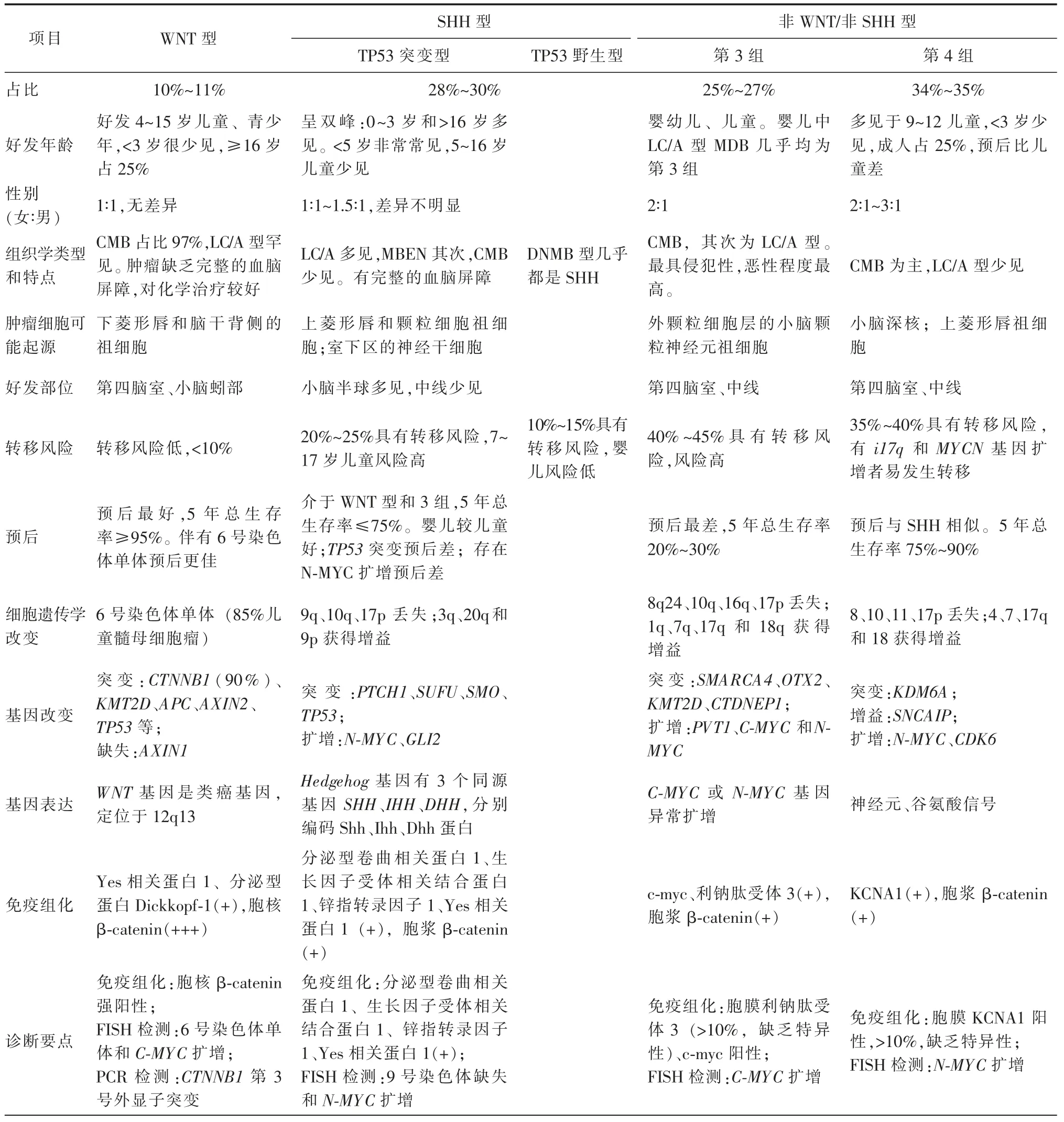
表2 髓母细胞瘤分子分型(基因分型)及亚型的特点
3.非WNT/非SHH型:最终表达Otx2,但缺乏其他标志物,如Yap1和核β-catenin。大多数非WNT/非SHH MDB可通过表达或甲基化谱进一步细分为临时的第3组和第4组变异体。MYC扩增被认为是非WNT/非SHH型MDB的一个重要预后生物标志物,MYC基因扩增可导致预后不良[12-13]。第3组包括标准风险MDB以及MYC扩增肿瘤,约占全部MDB的25%,组织学类型主要为CMB和LC/A,主要见于10岁以下的婴儿和儿童,成人罕见,可能是成人缺乏该亚型起源的前体细胞。MYC基因的扩增、i17q是非WNT/非SHH型MDB的主要遗传学特征,并常伴肿瘤早期扩散转移,预后最差,OS<50%[14]。而无MYC扩增或i17q、术前无转移者预后相对较好,5年OS波动在为55%~67%。
第4组MDB占所有MDB的35%~40%,约占成人MDB 25%,与MYC扩增和等i17q有关,可发生于所有年龄阶段,总体预后中等[15]。该亚型MDB的主要组织学类型为CMB和LC/A。常见的染色体改变是MYCN、CDK6基因扩增。i17q在第4组MDB更为多见,约80%的病例可见,而在第3组中则较少发生,可用于这2个分子亚型的鉴别[7]。第4组的预后较WNT型和SHH型差,特别是成人MDB中更为显著。存在17号染色体扩增或11号染色体缺失的患者,有相对较好的预后;不伴有上述2项染色体异常且为M1~4的患者预后较差。
(三)危险度分级
危险度分级与患者的治疗模式及预后密切相关,传统的危险度分级是结合MRI和脑脊液细胞学检查将MDB分为标准风险或高风险组。分级的主要依据是:有无蛛网膜下腔转移、患者的年龄、术后残留的大小。年龄>3岁、无转移性疾病(M0)、术后残留肿瘤<1.5 cm2、组织学上无间变性的儿童被归类为标准风险,其余的为高风险[16]。Ellison等[17]提出最新的危险度分级标准,除了低度危险组、高度危险组,还有中度危险组(59%),为非以上类型的患者(表3)。

表3 髓母细胞瘤的危险度分级
MDB的4种亚型具有显著异质性,具有不同的起源、好发解剖部位、人群分布以及预后和治疗选择。基于分子表型的危险性分层更为准确,有助于指导临床治疗和确定临床预后。目前,迫切需要建立基于分子生物学特征的危险分级,并开展个体化治疗策略。危险分层治疗是制定MDB治疗方案的主要依据,未来发展的方向是个体化治疗。
(四)制定治疗策略
术后需要根据MDB的不同危险分级,采用不同的综合治疗方案。Thompson等[18]对787例MDB患者利用分子分型(WNT型86例、SHH型242例、第3组163例、第4组296例),发现WNT、SHH和第3组肿瘤患者更大程度的切除范围对生存效益没有明显的影响,对于第4组MDB患者,与次全切除相比,完全切除可提高无进展生存率,尤其是转移性疾病患者。根据分子分型,研究针对每个分子亚型的靶向治疗,以便更好地个性化治疗。M1~4期意味MDB有着不良的生物学特性,需要采取更激进的治疗策略。低危组的全脑、脊髓放射治疗剂量只需要23.4 Gy,而高危组的放射治疗则需要36.0 Gy。Tarbell等[19]报道214例高危组MDB患者,采用全脑、脊髓轴40 Gy的放射治疗剂量,同时给予药物化学治疗,患者的5年无进展生存期和OS均在65%以上。Gandola等[20]采用超分割放射治疗(全脑脊髓轴为39 Gy,后颅窝为60 Gy)联合化学治疗,5年无进展生存期和OS分别为72%、73%,说明高危组患者需要更多的治疗模式以改善目前较低的生存率。
综上所述,正确认识MDB的分期和风险分级有助于指导制定恰当的治疗方案,提高疗效。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
基层中医药(2020年5期)2020-09-11 06:32:00
基层中医药(2018年5期)2018-08-31 02:35:42
中国卫生标准管理(2015年15期)2016-01-15 02:58:43
中外医疗(2015年11期)2016-01-04 03:58:43
中国病理生理杂志(2015年8期)2015-12-21 12:38:10
罕少疾病杂志(2015年1期)2015-07-12 08:51:30
制造技术与机床(2015年10期)2015-04-09 07:06:14
中国当代医药(2015年30期)2015-03-01 02:08:19
癌变·畸变·突变(2014年2期)2014-03-01 04:39:41
中国中医药现代远程教育(2014年21期)2014-03-01 04:32:23
