
肾性骨病相关分子通路的研究进展
2020-03-26 03:56:50伍子贤戴如璋林少豪冯元蓝梁德江晓兵任辉唐晶晶
中国骨质疏松杂志 2020年1期
伍子贤 戴如璋 林少豪 冯元蓝 梁德 江晓兵 任辉 唐晶晶
1.广州中医药大学,广东 广州 510405 2.广州中医药大学第一附属医院,广东 广州 510405
慢性肾病(chronic kidney disease,CKD)常继发于慢性肾炎、急性肾衰等肾性疾病,定义为任何持续时间超过3个月的肾脏结构和功能异常疾病[1]。相关调查显示,我国40岁以上人群慢性肾病的患病率大于10%,并且随着糖尿病发病率的上升,继发性CKD患病率不断上升[2],CKD人群不断扩大,该疾病严重影响患者的生活质量,每年CKD及其并发症导致的经济问题与社会问题越来越多,后者尤为突出。而CKD的主要并发症有肾性脑病、胃肠道异常、骨病等,其中,最常见的为肾性骨病(CKD-multiple myeloma bone disease,CKD-MBD)[3]。肾性骨病是CKD引发的一系列钙磷代谢紊乱和成骨-破骨异常而导致的一种疾病,是CKD引发矿物质及骨骼代谢失调的一种表现形式。根据病例特点分为高转运性骨病、低转运性骨病及混合性骨病三类[4-6]。
肾性骨病在慢性肾病人群中十分普遍,在一项对2 096例肾脏替代治疗患者的回顾性分析表明,在肾替代治疗人群中,血液透析患者的症状性骨折风险是肾移植患者的两倍,而腹膜透析患者的骨折风险介于两者之间,CKD患者未经透析的骨折风险可能更大[7]。故肾性骨病的发生大大降低肾病患者的生活质量且增加其死亡率。近年来,随着对肾性骨病关注度的不断提高,不仅进一步更完善地阐明了肾脏-PTH信号通路在肾性骨病的作用机制,也发现在肾性骨病的发病进展中有其他重要的分子信号通路参与调控,共同形成复杂的分子信号通路网。因此,深入探讨“分子信号通路生物学机制与分子信号通路网之间的关系”对于研究CKD发病机制及临床肾性骨病的防治具有一定的参考价值。
1 肾性骨病中与骨-肾轴相关的分子信号通路
研究发现,肾性骨病的患者会出现骨代谢营养疾病,继而导致骨吸收与骨形成平衡的紊乱。同时骨作为一种特别的内分泌器官,也能反作用于肾脏调控特定的分子信号通路。这种相互调控、相互影响的关系,被称为骨-肾轴[3,8]。
肾性骨病通过分子信号通路网络调控骨-肾轴,其中涉及的信号通路主要包括肾脏-PTH通路(轴心通路)、Wnt/β-catenin通路和骨形态发生蛋白(bone morphogenetic proteins,BMP)通路,后两者作为肾脏-PTH通路下游通路,3个通路共同协助调节骨形成与骨吸收;klotho基因平衡协助通路与轴心之间的关系;骨FGF23分子反馈性调节轴心通路(见图1)。当肾脏疾病时,肾性骨病通过上述信号通路网引起该病的发生。
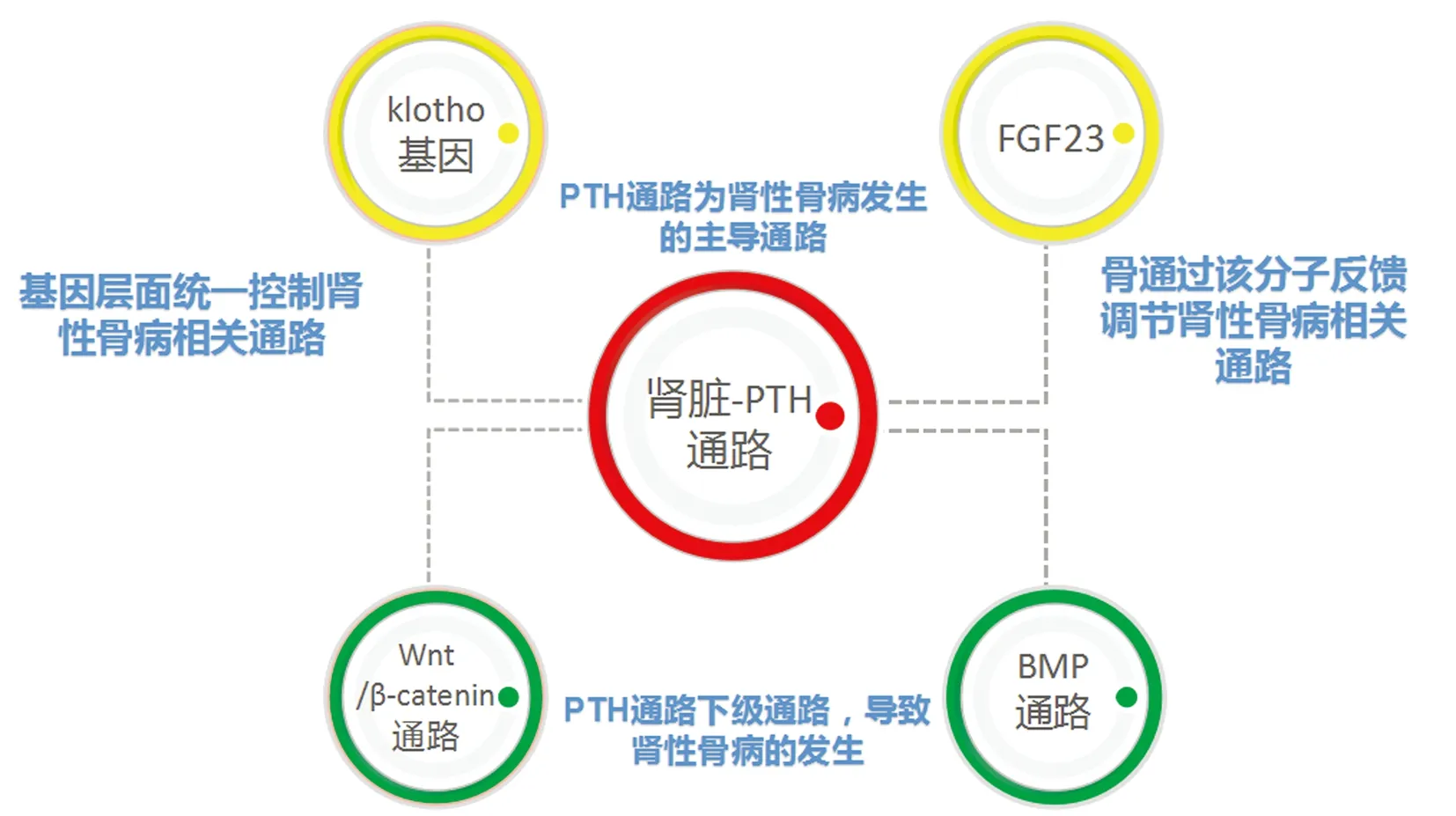
图1 肾性骨病中肾-骨轴的分子信号通路网Fig.1 Molecular signaling pathway network of renal-bone axis in renal osteodystrophy
1.1 肾脏-PTH通路为肾性骨病骨-肾轴的轴心
在正常情况下,由甲状旁腺分泌的PTH所介导的通路能够通过调节骨钙与血钙的平衡来调控骨平衡,是参与骨合成代谢的重要信号通路。其主要的通路机制为:PTH主要与PTH 受体(PTHR)结合而发挥其生物功能。PTH1R是II型G蛋白偶联受体,具有2条信号通路[9]:(1)Gsɑ偶联的依赖cAMP的蛋白激酶A信号途径;(2)Gq/11偶联的PLC-蛋白激酶C信号途径。CKD继发钙磷代谢紊乱甲状旁腺增大,PTH分泌过多,最终导致骨平衡彻底紊乱。见图2。
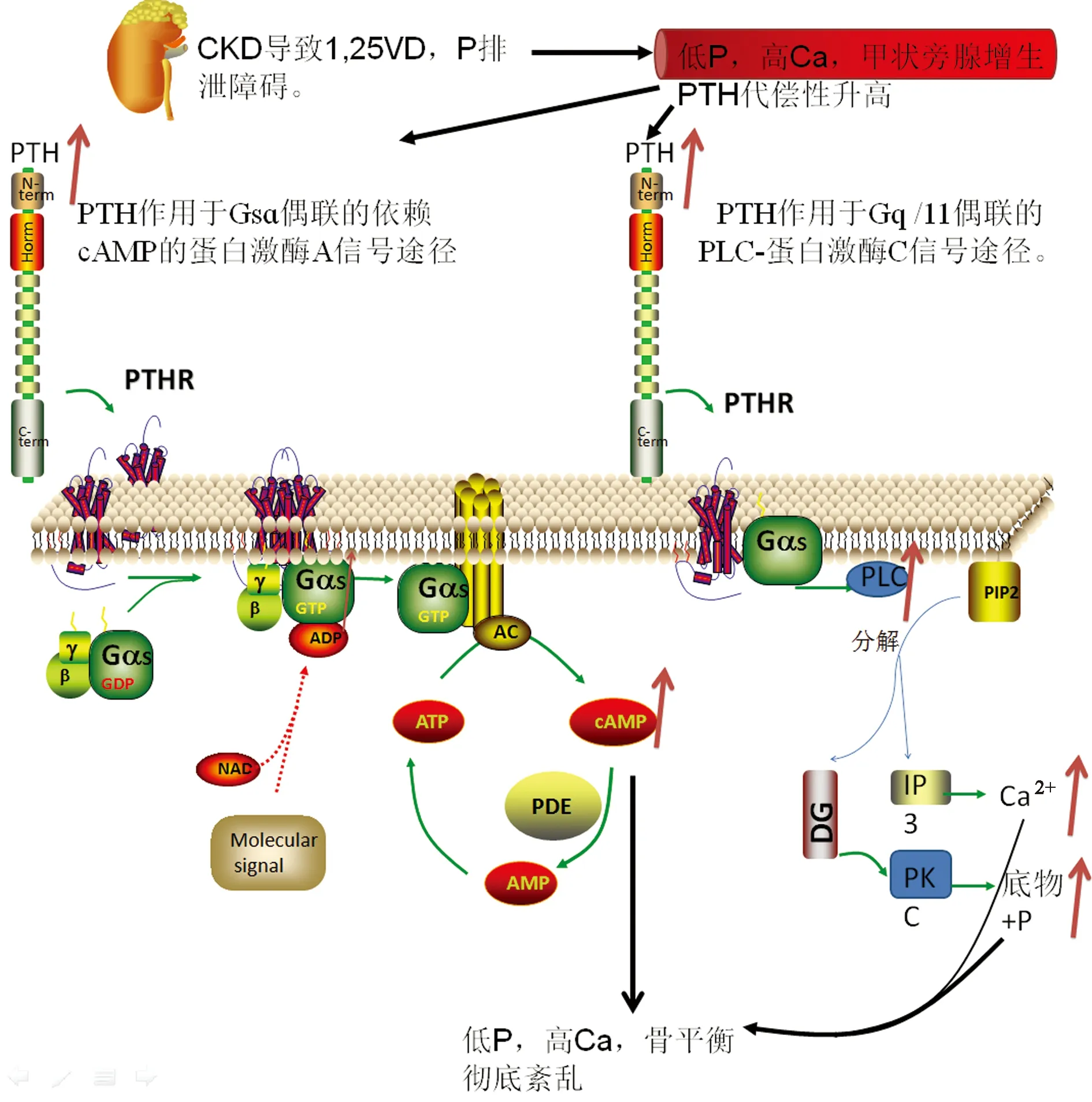
图2 PTH1R 的两条信号通路Fig.2 Two signaling pathways of PTH1R
现代医学认为[10-11],CKD-MBD主要是由于肾功能下降导致钙的代谢排泄受到影响,故肾脏生成1,25(OH)2D3的能力下降,同时肾脏排泄磷离子能力下降,导致血清磷的水平过高,钙代谢紊乱,引发一系列机体钙磷平衡的紊乱,继而导致了甲状旁腺增生,异常分泌PTH增多,此过程可称为继发性甲状旁腺功能亢进。血清PTH水平过高,生物通路机制紊乱,破骨细胞活性代偿性增加,成骨细胞活性代偿性下降,释放骨钙入血,可单独或同时导致成骨-破骨过程异常,最终表现为矿物质、1,25(OH)2D3代谢异常,骨转化、矿化、容量、线性生长或强度异常以及血管或软组织异位钙化[3]。故CKD晚期的患者几乎都会存在CKD-MBD的问题,严重的可导致骨质疏松和骨折等严重并发症的发生,极大地降低CKD患者的生活质量,缩短患者的生存时间。
在临床研究中,针对肾脏-PTH通路分子靶点进行相应的用药,能够在临床上取得确切的疗效,如:给予1,25(OH)2D3与钙磷结合剂[12],或者调节透析的方式[13]清除异常的钙磷代谢物,能够大大缓解肾性骨病的病程进展;现存最新的治疗方案是对CKD患者早期运用甲状旁腺切除术并甲状旁腺移植术[14],该方案能取得一定的疗效[3]。但在最新的研究发现,单纯的针对肾脏-PTH通路进行用药治疗效果已经不能满足越来越高的医疗需求,同时调控与轴心相关的分子信号通路提高预后已成为最新的治疗观念。
1.2 Wnt/β-catenin通路
正常情况下,Wnt/β-catenin通路作用机制如下: Wnt蛋白与细胞膜受体(LRP5/6)结合,活化Fz受体,通过骨架蛋白(Dv)和酪蛋白激酶1(CK1)传递信号,活化由轴蛋白(axin)、结肠腺瘤性息肉病蛋白(APC)和糖原合成酶激酶3(GSK3)组成的复合物,激活细胞内信号通路[15-16]。无Wnt信号刺激时,β-catenin被GSK3磷酸化,并通过泛素/蛋白酶体途径被降解。而当有Wnt 信号刺激时,蛋白复合物解离不发生磷酸化,catenin在胞浆中积聚并转位入核,与转录因子T细胞因子/淋巴细胞增强因子(TCF/LEF)结合启动靶基因如c-myc、cyclin D1的转录,促进细胞的增殖或活化,该过程对成骨非常关键[17](见图3)。
此过程可受多种调节因子共同调节,其中硬化蛋白(sclerostin,SOST)与 Dickkopf 1(DKK1)是目前被研究最多的,也是最为重要的 Wnt/β-catenin 信号通路的两个抑制物,其作用是主要通过与LRP5/6共受体结合阻碍具有活性的Wnt受体复合物的形成[18],从而抑制Wnt/β-catenin信号的转导(见图3)。高表达的SOST主要通过抑制 Wnt/β-catenin信号通路的激活,阻碍间充质干细胞向成熟的骨细胞分化,降低骨形成,调节骨量[19]。DKK-1是一种糖蛋白,在胚胎形成过程中表达于多种组织中(包括骨组织),通过与LRP6以及跨膜蛋白Kremen1/2形成复合体,导致 LRP内吞与降解,减少细胞膜上可用性的LRP与Wnt配体结合[20-21],阻断了Wnt信号向胞内的传递,从而抑制骨形成,降低骨量[22]。
近期研究显示,wingless related MMTV integration site(Wnt)/β-catenin 信号通路在肾性骨病发病过程中具有至关重要的作用(见图4),PTH能够使GSK3β失活[23],从而稳定β-catenin[24]。另外,PTH与PTH1R结合后,PTH1R与LRP6形成复合物,使得具有破坏性的复合物(即由轴蛋白、APC和GSK 3组成的复合物)的解离[24],从而避免β-catenin被降解而激活了Wnt信号途径。PTH 能够作用于骨细胞以减少Wnt抑制因子骨硬化蛋白[25]和DKK1的表达[26],这些内源性抑制因子的减少导致了自分泌Wnt信号途径的增强,且在动物实验中,PTH对敲除LRP5基因的小鼠并不发挥其促骨合成代谢的作用[25,27],提示PTH通路对Wnt下游信号通路的作用。
综上,在肾性骨病中Wnt/β-catenin通路作为肾脏-PTH通路的下游通路,PTH可通过对硬化蛋白(sclerostin,SOST)、Dickkopf 1(DKK1)和β-catenin的调控,控制Wnt/β-catenin通路激活与否,进而影响肾性骨病的进展。
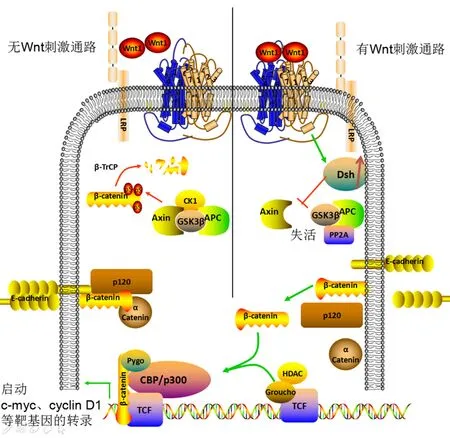
图3 正常的Wnt/β-catenin信号通路Fig.3 Normal Wnt/β-catenin signaling pathway
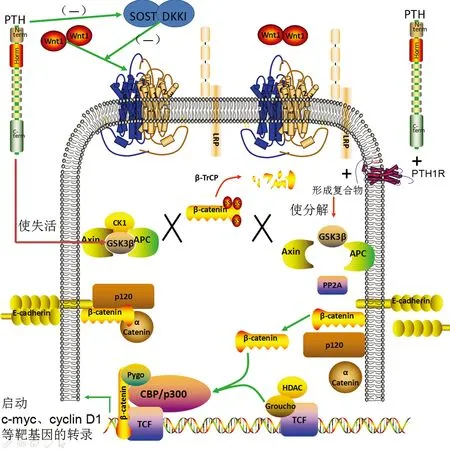
图4 PTH与Wnt/β-catenin通路作用机制Fig.4 Mechanism of action of PTH and Wnt/β-catenin pathway
PTH通过抑制SOST,DKK1与复合体中β链蛋白的活性调节Wnt/β-catenin通路的激活与否。同时,PTH与PTH1R结合激活PTH1R,PTH1R与LRP蛋白结合形成复合物,可促使由轴蛋白(axin)、结肠腺瘤性息肉病蛋白(APC)和糖原合成酶激酶3(GSK3)等组合成的复合物分解,使细胞内β-catenin的含量上升。
1.3 BMP通路
BMP属于转移生长因子TGF-β超家族中的一员,其主要生物学作用是诱导未分化的间充质细胞增殖及发生成骨分化,最终促进骨的形成。在CKD-MBD中,研究发现随着PTH信号通路异常,肾性骨病的病程进展,血清中的BMP-7会持续下降,说明PTH信号通路的异常可能导致了BMP通路的功能下降[28],从而加速了肾性骨病的进展。
BMP主要信号通路是通过与相应的受体结合,激活细胞内下游通路如Smad和MAPK等信号通路,从而发生一系列磷酸化或聚合酶联反应[29]。其中与肾性骨病最为相关的分子为BMP-7[30]。在实验研究中,一方面BMP-7可以逆转肾纤维化进程,诱导间充质细胞向上皮细胞的转化,保护肾功能,促进损伤肾再生修复[30];另一方面在慢性肾性骨病模型的动物实验中[31],给予外源性的BMP-7可以使血磷显著降低,骨质病变基本消失;除此之外,在细胞层面上,还可以增加成骨细胞活性,抑制骨小梁纤维化,减少骨的重吸收。同时,BMP-7还能在一定程度上预防肾性骨病血清钙异常在血管内膜沉积而导致血管硬化的严重并发症,避免发生血管疾病。
1.4 Klotho基因
日本一个研究衰老的研究团队发现机体中存在一种名为Klotho基因的抗衰老基因,它能够协调肾性骨病相关的分子信号通路。Klotho基因表达蛋白有两类,即循环型Klotho蛋白(Circulating Klotho)与跨膜型Klotho蛋白(Trans-membrane Klotho)[16]。该表达蛋白可以作为一种新型的去糖基类固醇类b型葡萄糖醛酸酶和钙通道瞬变受体电位vallinoid-5(TRPV5)发挥相应的生理功能;或作为FGF23刺激成纤维细胞生长因子(FGF)受体的辅助因子。Klotho基因表达蛋白的这两项功能导致了Klotho基因成为调节矿物质和维生素D代谢的关键因子[32],使之可以在肾脏功能中发挥重要的调控作用。Klotho基因的表达受到雌激素、钙、磷酸盐、1,25(OH)2D3、PTH和年龄等的影响,其表达会受到影响,从而维持上述分子的动态平衡,以保护骨-肾结构;而当Klotho基因表达异常的时候,反而会加速肾性骨病的进展[16]。
在相关动物模型实验研究中[33-35],对于1,25(OH)2D3的调控,当小鼠Klotho基因高表达时,小鼠内环境出现钙磷酸盐稳态紊乱和血清活性维生素D[1,25(OH)2D3]浓度异常升高的情况;当这些小鼠模型大多数发展为衰老表型(包括肾-骨损伤),即衰老致Klotho基因表达水平下降,该内环境紊乱可一定程度上被纠正,可见血清钙和1,25(OH)2D3浓度降低。同时通过饮食中磷酸盐的限制[17-18],提示这些表型是下游事件。有研究者推测,Klotho基因异常表达模型可能通过调节25-羟维生素d-1a-羟化酶(CYP27b1)活性调节1,25(OH)2D3,但这种机制具体尚不清楚。
另外,通过对Klotho基因突变转基因小鼠的研究发现,突变体小鼠会出现高钙血症或高磷血症。其中一种途径近期已被阐明[33-35]:Klotho基因通过一种新的分子机制调节,即通过细胞膜表面的去糖基化和稳定上皮钙通道TRPV5实现远曲小管中钙的再吸收。Klotho基因、TRPV5和 calbindin-D28 K在小鼠肾细胞远曲连接小管中结合并发挥其功能,使肾段负责活跃的跨上皮钙再吸收的相应小管功能增强。相反,缺乏Klotho基因会导致细胞表面TRPV5的表达减少,减少管状钙的再吸收。同样,缺乏TRPV5的小鼠减少了Klotho基因表达,并降低了肾钙的再吸收,尽管增加了1,25(OH)2D3的水平,但并不能维持原有的平衡。
除此之外,由于高钙血症、高维生素D及Klotho基因对磷酸盐的肠道和肾吸收的影响,Klotho基因表达蛋白的缺乏还可能会减少尿中磷酸盐的排泄导致高磷血症的发生。
1.5 FGF23分子相关的骨反馈机制
FGF23是一种主要由骨细胞分泌的32-kDa蛋白(含251个氨基酸)。FGF23通过与肾脏中的特定受体相互作用,可以反馈调控肾的相关分子信号通路(BMP通路、Klotho基因等),从而调节肾脏磷酸盐排泄和维生素D合成,在肾-骨轴机制中扮演了一个重要的新角色[8]。
这种骨激素需要特定的辅助因子能够有效地激活其受体。这类辅助因子主要以Klotho基因为代表,已知在男性体内主要在肾脏产生,为现研究中较为热点的骨-肾模型。而在最新的研究进展中[8],其中两个辅助因子已经被发现:第一个被发现的辅助因子Klotho[16]能与受体FGFR1c(FGF23受体)结合,并激活该受体;而第二因子b-Klotho[21]与肝脏、胰腺和脂肪细胞中剩余的FGF受体相互作用,最终改变维生素D、钙和磷酸盐的平衡[36],甚至影响胆汁酸和能量的稳态。基于上述Klotho基因与FGF23的相互作用,相关研究者通过动物实验进一步证实:FGF23与Klotho基因存在重要联系[21],因此可以推断出FGF23与骨病之间存在重要联系,但背后的机制需要进一步阐明。
基于临床观察与特殊病例的收集(严重低磷酸酯血症相关的肿瘤),有研究者已经提出关于FGF23作用的假设——骨-肾轴简化方案[21,36](见图5)。一方面,由骨细胞产生的FGF23影响肾脏活化维生素D的合成和磷酸盐排泄。同时,FGF23活性直接或间接受到骨源蛋白的影响。特别是由细胞外基质磷酸糖蛋白(MEPE)的裂解而产生的酸性丝氨酸、天冬氨酸富集序列(ASARM),具有磷酸酶-尿的作用。同时,牙本质基质酸性磷蛋白1(DMP 1)通过刺激X连锁磷酸盐调节基因(PHEX),间接降低FGF23活性。另一方面,血清中磷酸盐和(或)1,25(OH)2D3水平的增加刺激骨细胞产生FGF23。由于Klotho基因在肾脏中的表达,FGF23可以结合其在肾小管细胞上的受体。从而抑制了Na-Pi 2a共转运蛋白的合成,增加了肾小管细胞的内吞作用,最终减少肾磷酸盐的再吸收和血清磷酸盐的含量。FGF23受体的激活还能抑制1-羟化酶,增加24,25-羟化酶的合成,从而减少活化维生素D的生成。建立了一个清晰的反馈回路,二者构成现有骨-肾之间的联系。
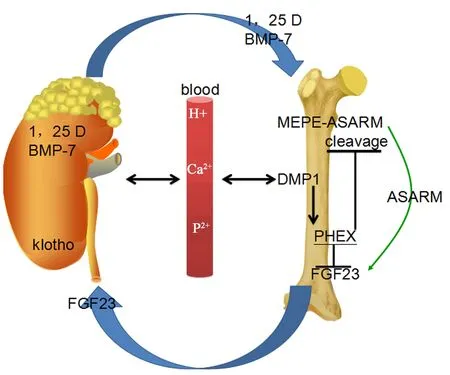
图5 骨-肾轴简化方案Fig.5 Simplified schema chart of the bone-kidney axis
除了胆甾醇外,肾源性BMP-7也可以影响骨细胞的活性。另一方面,FGF23是由骨细胞产生的,它会影响肾脏活化维生素D(骨化三醇)的合成和磷酸盐的排泄。同时,FGF23活性直接或间接地受到骨源性蛋白的影响。特别是由于MEPE的裂解而产生的ASARM具有磷-氮效应,DMP1通过刺激PHEX间接降低FGF23的活性。骨与肾之间的激素对话能调控血液电解质稳态。
2 结语
寻找肾性骨病相关分子信号通路上的作用靶点并进行相关研究,是制定合理有效的肾性骨病治疗方案的重要途径。目前研究中,肾性骨病主要涉及的分子信号通路有:以肾脏-PTH通路作为肾性骨病中调控骨-肾的轴心通路,Wnt/β-catenin通路和BMP通路作为肾脏-PTH通路下游通路,共同协助调节骨形成与骨吸收;Klotho基因平衡协助通路与轴心之间的关系;骨FGF23分子反馈性调节轴心通路。
本文就其研究广度而言,除了上述所报道的分子信号通路以外,相关文献还报道了雌激素通路、谷氨酸通路、OPG/RANKL/RANK信号通路参与在分子信号通路网中,但上述通路详细机制尚不清楚,仍有进一步研究的必要。
通过临床观察与临床治疗发现,在当前临床治疗大部分药物单纯针对肾脏-PTH通路且作用靶点单一,或仅纠正PTH通路异常所引发的微量元素紊乱,此类用药更多用于早期肾性骨病的预防与治疗上,对于肾性骨病后期所导致的骨疾病尚且无较好的治疗效果。故今后有必要进行相关药物的研究,通过调控与轴心相关的分子信号通路提高预后效果,同时兼顾肾性骨病后期导致的骨疾病,以建立更有效的治疗方案。
猜你喜欢
中国民间疗法(2021年19期)2021-11-20 06:22:34
中国民间疗法(2021年18期)2021-11-02 08:20:16
疯狂英语·新读写(2018年3期)2018-11-29 22:37:11
现代营销(创富信息版)(2018年10期)2018-10-12 03:01:26
现代营销(创富信息版)(2018年8期)2018-09-08 08:51:56
现代营销(创富信息版)(2018年9期)2018-09-03 09:49:34
中成药(2017年8期)2017-11-22 03:19:29
电镀与环保(2016年2期)2017-01-20 08:15:25
中国继续医学教育(2015年4期)2016-01-07 07:38:10
中国合理用药探索(2014年11期)2014-03-11 20:30:22
