
常染色体隐性遗传性多囊肾合并多囊肝1例报告
2020-03-26 07:41李嘉林
临床肝胆病杂志 2020年3期
李嘉林, 陈 铭
1 广州中医药大学, 广州 510000; 2 广州中医药大学第一附属医院 泌尿外科, 广州 510000
多囊肾是一种常见的单基因先天性遗传病,可以显性或隐性方式遗传[1]。其中,常染色体隐性遗传性多囊肾(autosomal recessive polycystic kidney disease, ARPKD)较为罕见,通常被认为是由PKHD1突变引起的遗传同质性疾病,与纤毛功能障碍有关,发病率约为 1∶20 000,多见于婴幼儿时期,存活至成人者极少[2],至今国内外仍少有报道。其特征在于肾集合管的非阻塞性梭形扩张和肝脏的导管板畸形,导致进行性慢性肾病和肝脏疾病,包括扩张的胆管、先天性肝纤维化和门静脉高压症(Caroli综合征)[3-5]。现将收治的1例成年ARPKD女性病例报道如下。
1 病例资料
患者女性,43岁,因“反复乏力、纳差2年,下肢水肿1周”于2019年2月1日就诊于广州中医药大学第一附属医院。患者2年前因乏力、恶心、纳差查血肌酐325 μmol/L,彩超结果提示多囊肾、多囊肝(具体不详),予对症处理后病情较前缓解,后反复于门诊治疗,肌酐呈进行性升高。1周前因劳累后乏力加重,伴胸闷气促及双下肢水肿症状,遂拟“多囊肾”收入院。入院见患者双下肢水肿,久坐、行走后症状明显,乏力,无胸闷气促等不适。既往史:高血压1年余,最高达170/100 mm Hg,诉规律服用硝苯地平控释片30 mg,2次/d,血压控制在130/80 mm Hg左右。家族中父母、兄弟姐妹、子女皆无多囊肾、多囊肝病史。查体:腹膨隆,轻微压痛,无反跳痛,腹部无包块。触诊肋下可触及肝脏约8 cm,脾脏肋下未触及,Murphy征阴性,肝区无叩击痛,无移动性浊音。双肾未触及,双肾区轻叩击痛,双侧输尿管行程无压痛,双下肢中度凹陷性水肿。入院后生化检查:白蛋白35.8 g/L,血肌酐517 μmol/L,尿素氮15.39 mmol/L。双肾彩超示:多囊肾并双肾多发结石(图1)。肝胆脾胰彩超示:多囊肝,腹腔积液(图2)。诊断为:(1)多囊肾(ARPKD);(2)肾终末期疾病,血液透析;(3)多囊肝;(4)高血压2级(很高危组)。完善相关检查,于局麻下行左上肢动静脉瘘成形术,定期行血液透析治疗(每周一、周四);药物治疗上予以控制血压、护肾、护肝等对症处理后,患者于2019年4月10日症状好转出院。出院后定期在本院门诊就诊及电话随访,目前一般情况良好。
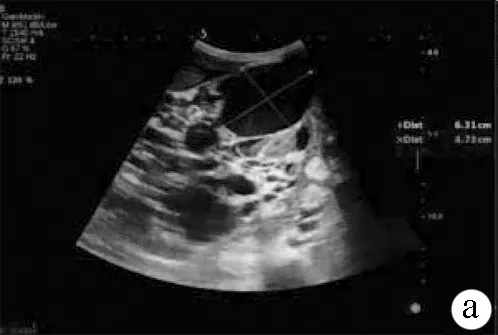
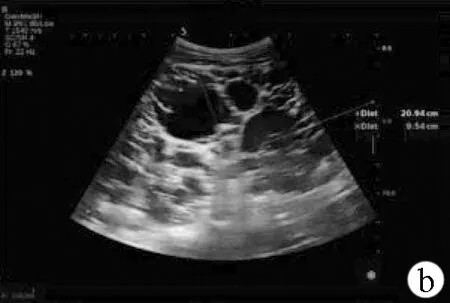
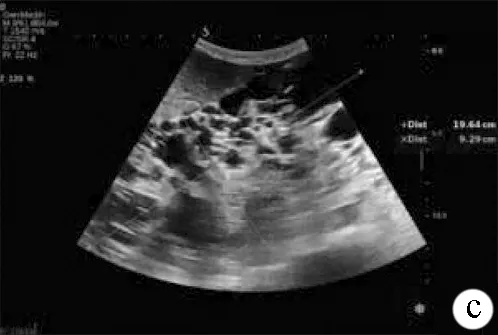
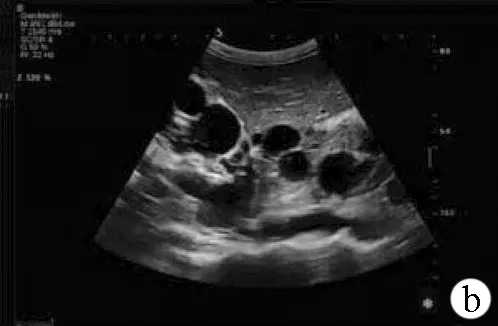
注:a,肾脏被大小不等的囊性结构取代,其中囊性组织较大者约63 mm×47 mm;b、c,双侧肾脏显著增大,左肾209 mm×95 mm,右肾200 mm×93 mm,肾包膜不规则,表面凹凸不平,有结节状隆起,肾脏内部结构不清,无法分辨肾皮髓质和肾窦的关系,未见正常肾实质回声。
图1患者肾脏彩超声像
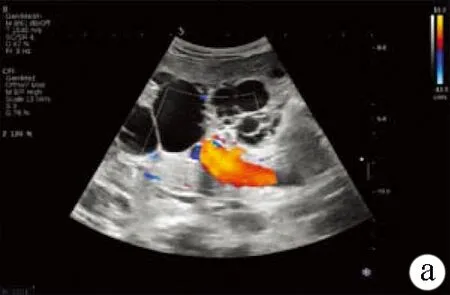
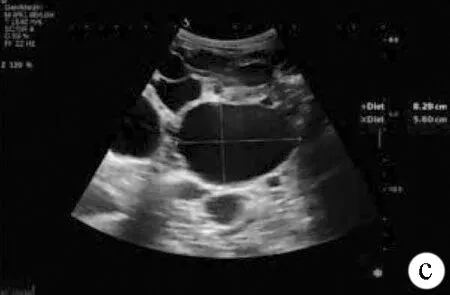
注:a(多普勒彩超成像)、b(普通B超成像),肝脏体积明显增大,形态失常,以右肝明显,肝脏面凹凸不平,肝内密布大小不等的类圆形囊性无回声区;c(普通B超成像),肝脏囊性组织较大者位于右叶,约82 mm×56 mm,无回声区间未见明显正常肝组织。
图2患者肝脏彩超声像
2 讨论
ARPKD属于一组先天性肝肾纤维囊性综合征,是儿童肾脏相关发病率和病死率的重要原因[6]。该病发病机制尚不明确,一般认为是由PKHD1突变引起,目前PKHD1基因突变方式已报道300余种[7]。PKHD1是人类基因组中最大的疾病基因之一,其基因组长度至少为470 kb,包括86个外显子,该基因位于第6号染色体上,位置为6P21.1-P12,ARPKD则是PKHD1基因突变导致的一种早发性肝肾纤维囊性疾病[8]。近几年亦有学者[9]通过对小鼠和斑马鱼的功能丧失研究进而提出DZIP1L可能是ARPKD发病机制的第二个基因。该病患者在孕检或刚出生时即可被确诊,25%~30%被确诊的新生儿出生不久后即可死亡[10],幸存者症状可呈进行性加重趋势,并由此引发其他疾病,包括高血压、肾衰竭、门静脉高压、肾和肝纤维化等。所有ARPKD患者都存在肝脏相关疾病[6]。相关研究[11]证明,所有患有ARPKD的患者在出生时均有胆管异常,其主要病理标志是由于原发性导管板畸形伴有周围纤维化导致胆管发育不全,最终发展为先天性肝纤维化和肝内胆管扩张(Caroli病)等。此类患者主要临床表现有脾大、腹胀腹痛、胃食管反流、血小板计数低、胃食管静脉曲张伴有急性出血等症状。
目前用于诊断ARPKD的检查主要包括彩超、CT和MRI等,有研究[4]表明超声声脉冲辐射力肝脏和脾脏弹性成像是检测和量化ARPKD患儿肝纤维化和门静脉高压有用的非侵入性生物标志物。CT检测较超声更为敏感且精准,但由于价格昂贵且辐射较强,故应用不甚广泛。MRI敏感度强,无电离辐射,较CT更为清晰,在未来可能成为诊疗ARPKD的主要非侵入性影像诊断工具。相较于CT、MRI,彩超因其易用性、低成本,仍是目前最为广泛应用的检查手段。该患者入院后查双肾彩超提示:双侧肾脏显著增大,肾包膜不规则,表面凸凹不平,有结节状隆起,肾脏内部结构不清,无法分辨肾皮髓质和肾窦的关系,未见正常肾实质回声,被大小不等的囊性结构取代。且患者父母、兄弟姐妹无肾囊肿疾病史,根据Zerres等[12]提出的修改标准,可诊断为ARPKD。
ARPKD目前治疗主要以缓解临床症状为主,暂无有效方法延缓病情进展。一般治疗通过改善患者生活习惯以减少对肝肾功能的过度损害;并积极防治并发症,如高血压、泌尿系感染、电解质紊乱、急性心肝肾等器官功能性损伤等,注意尽可能减少药物使用不当引起的肝肾功能损伤增加;早中期ARPKD患者可选择外科手术,手术治疗可明显减轻患者临床症状,但对肾功能改善不明显;晚期患者须行肾脏替代疗法,主要以血液透析为主,必要时可行肾脏移植术。对于合并多囊肝的患者,治疗过程中应注意对肝功能的监测,目前多囊肝的治疗以外科手术为主,主要术式包括肝动脉栓塞术、肝部分切除术、肝脏移植术等,可有效缓解患者临床症状、改善患者生活质量,但远期效果不理想[13]。
成年人ARPKD合并多囊肝病例在临床上极为罕见,尽早确诊、干预可在一定程度上提高患者生存率。目前尚无有效治疗手段,主要以积极处理并发症为主。近年来,随着人们对ARPKD病理生理学认识的不断深入,相继出现了许多针对分子基因遗传学方面的治疗研究,虽然目前暂时缺乏大数据观察及强说服力的证据,但相信在不久的将来,人类在该病的治疗上会有大的突破。
猜你喜欢
家庭医药(2021年2期)2021-03-09
保健医苑(2020年6期)2020-12-04
基层中医药(2020年12期)2020-07-22
中国临床医学影像杂志(2019年5期)2019-08-27
中国临床医学影像杂志(2019年1期)2019-04-25
心肺血管病杂志(2019年1期)2019-04-22
中华老年多器官疾病杂志(2016年7期)2016-04-28
癌变·畸变·突变(2015年3期)2015-02-27
中国中医药现代远程教育(2014年23期)2014-03-01
中国全科医学(2014年14期)2014-01-28
