
Screening ulcerative colitis key target gene and pathway based on KEGG pathway
2020-03-14 04:38:06ZhiHuaYangYanLiuYiHuaFanHaiFengYanFirstTeachingHospitalofTianjinUniversityofTraditionalChineseMedicineTianjin30038ChinaTianjinNankaiHospitalTianjin30000China
Precision Medicine Research 2020年1期
Zhi-Hua Yang, Yan Liu, Yi-Hua Fan, Hai-Feng Yan*First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, Tianjin 30038, China. Tianjin Nankai Hospital,Tianjin 30000, China.
Abstract Objective: To obtain the relevant information of pathway and gene by using the database of DAVID, analyze the function and distribution of important genes, screen out the target genes related to Ulcerative Colitis and promote the study of the pathogenesis of UC and the development of new drugs. Methods: The Ulcerative Colitis was used to search UC related genes in TTD, Drugbank, DisGeNET database. The obtained gene data was input to the database of Daved, and the data of gene enriched pathway was obtained 87 genes were Significant enriched in the first 20 KEGG pathways. The 87 genes were input to the string database to make the interaction network diagram,and the key genes enriched in the pathway were also made the network diagram, and the two network diagrams were compared. Results: RELA, TNF, IL1B, NFKB1, IL6 and IL10 were among the highest ranked genes in two network diagrams. Conclusion: The study of RELA, TNF, IL1B, NFKB1, IL6 and IL10 is necessary for us to study further. The pathogenesis of UC was associated with multiple pathways such as NF-kappa B signaling pathway, Jak-STAT signaling pathway, TNF signaling pathway and so on. It is helpful to understand the pathogenesis of disease and provide a reliable target for the development of new drugs by analyzing the pathway and the network diagram of the interaction between genes and disease related genes.
Keywords: Ulcerative Colitis, KEGG pathway, Target gene, Pathway
Background
Ulcerative colitis (UC), a type of inflammatory bowel disease, is a chronic idiopathic inflammatory bowel disease. It is characterized by continuous mucositis starting from the rectum and extending to the proximal side [1], clinical symptoms mainly including bloody stool, diarrhea and weight loss [2], with the characteristics of recurrent and chronic persistent [3].According to epidemiological studies, UC is more common in Asia, with a clear upward trend, and gender distribution tends to be equal. In addition, the rising incidence of UC may increase the risk of colorectal cancer [4]. At present, the pathogenesis and pathogenic factors of the disease have not been clarified, and it is a research hotspot in digestive system diseases. This study combines KEGG pathway and gene interaction network for comprehensive analysis to identify possible drug targets for UC and provide reliable targets for new drug development.
Research methods
Collecting UC related genes
Finally, 315 UC genes were identified by using database TTD 4.3.02 (http://db.idrblab.org/ttd/) [5],Drugbank 5.0 (http://www.drugbank.ca/) [6] and DisGeNET v5.0(http://www.disgenet.org/web/DisGeNET/menu/home)after deleted the false positive and duplicate genes [7].
KEGG pathway enrichment analysis
Different genes cooperate with each other to play corresponding biological functions in the body.Pathway-based analysis contributes to further understand the biological functions of genes. In this paper, David (version 6.8) was used to analyze the pathway of UC related targets. And the important pathway was selected for key analysis, which is conducive to understanding the pathogenesis of the disease, and also can provide a reliable target for the research of new drugs.
Interaction network analysis of genes significantly enriched in pathways
According to the results of DAVID database analysis,genes enriched in the first 20 pathways and genes appearing on the pathway with frequency ≥ 9 were input into the String database (http://string-db.org/) [8]to construct protein-protein interaction network model[9], and the species was set as "HOMA sapiens". To ensure the reliability of the data, the minimum protein interaction threshold was set to "highest confidence" (>0.9). The protein interaction was screened, the results were saved in TSV format, and the node1, node2, and combined score data in the file were retained and imported into Cytoscape 3.5.1 software for analysis of gene interaction network diagrams. Degrees of freedom and betweenness are two main topological parameters that measure the importance of a node in the network, and are also important references for determining whether a gene is a "core target" [10]. The greater the degree of freedom, the stronger the biological importance, and the greater the betweenness,the more important the node is in the network. The size of a node is determined by the degree of freedom, the greater the degree of freedom, the larger the node. The color of the node is set according to the degree of freedom, and the degree of freedom gradually increases as the color changing from blue to yellow.The thickness of the edge is determined by the betweenness, and the larger the betweenness, the thicker the edge. Compare the total gene network map enriched in the pathway with the gene network map with frequency of 9 or more to find out the genes that are shared by the two and have the highest degree of value, and these genes may be the targets of disease occurrence and drug treatment gene.
Enrichment analysis of Gene Ontology function
Gene Ontology (GO) is a gene function classification system that describes the functions of genes, genes and gene products in an organism, including molecular function (MF), biological process (BP) and cellular components (CC) 3 branches [11]. In this paper, P <0.01 was used as the screening condition and (DAVID,https//david.ncifcrf.gov/ summary.jsp, Version 6.8) was used to perform GO functional enrichment analysis on UC key genes.
Results
Screening of UC genes
34 UC genes were collected in the TTD database, 58 UC genes were collected in the Drugbank database,233 UC genes were collected in the DisGeNET database, 10 duplicate genes were removed, and 315 UC genes were finally collected.
DAVID pathway analysis
In the DAVID database, 315 related genes were input and their KEGG pathways were analyzed and 298 genes were enriched on 85 pathways. Based on P <0.01 as the screening condition, we analyzed the 20 pathways with the most significant enrichment. A total of 87 genes were annotated to these pathways, as shown in Table 1. 16 genes appeared in 20 pathways with frequency ≥ 9, as shown in Table 2.
Networkanalysis of enriched genes and high frequency genes
A total of 87 UC-related genes were significantly enriched in the first 20 pathways, and 16 genes appeared in 20 pathways with a frequency of ≥ 9.These 16 genes may be closely related to the pathogenesis of UC, because they are involved in the signaling of multiple pathways. In order to study the interaction between genes, 87 genes and 16 genes with high frequency were input into the string database to make the network map of gene interaction, and the genes without interaction were removed. The results of topology analysis were carried out by the software of Cytoscape 3.5.1, and the two network maps werecompared. In the graph, the degree and betweenness centrality of the nodes directly reflect the degree of association with the disease. The greater the degree of freedom, the stronger the biological importance, and the greater the betweenness centrality, the more important the node is in the network. Figure 1 is the 80 genes enriched in 20 pathways (7 genes are not shown in the figure), and Figure 2 is the 16 genes with frequency of 9 or more in 20 pathways. The genes with the highest degrees of nodes in the two graphs may be the target genes we are looking for. In Figure 1, there are 80 nodes and 304 interactions, and in Figure 2,there are 16 nodes and 47 interactions. There are six genes that are common and highly ranked in the two figure, including RELA, TNF, IL1B, NFKB1, IL6, and IL10, indicating that these six genes are closely related to UC and should be our target genes for UC research.
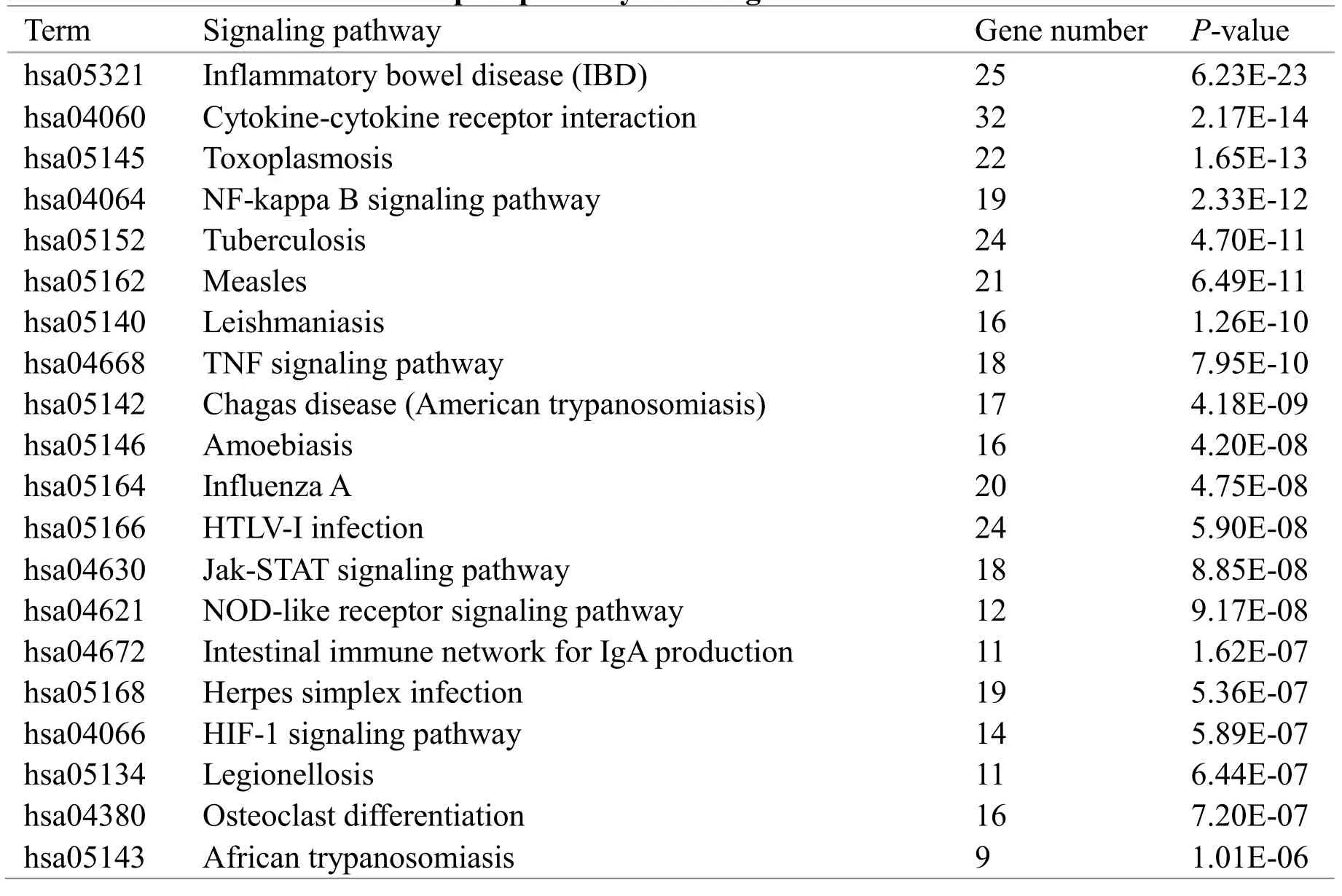
Table 1 Top 20 pathways with significant enrichment

Table 2 Genes with a frequency of ≥ 9 in 20 pathways significantly enriched
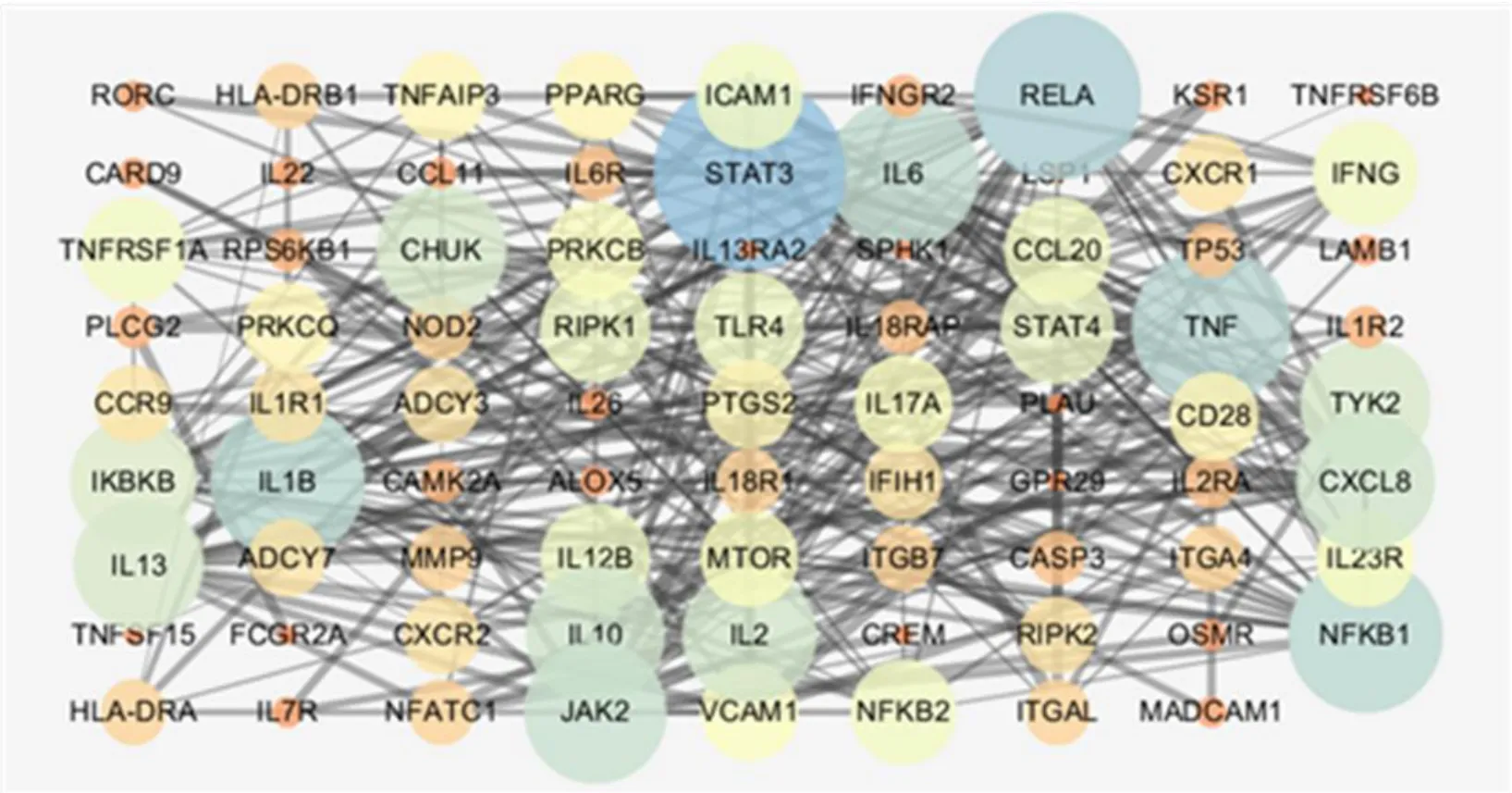
Figure 1 Network diagram of 80 gene interactions
Enrichment analysis of top-ranked gene functions
Genes with a frequency of ≥ 9 on 20 pathways that were significantly enriched were entered into the DAVID database for GO functional enrichment analysis. The top-ranking biological processes of genes were mainly concentrated in positive regulation of NF-kappa B transcription factor activity, cellular response to lipopolysaccharide, inflammatory response,immune response, etc. Cell components are mainly concentrated in the external side of plasma membrane,cell surface, extracellular space, etc. The molecular functions are mainly concentrated in cytokine activity,protein heterodimerization activity, etc., as shown in Table 3.
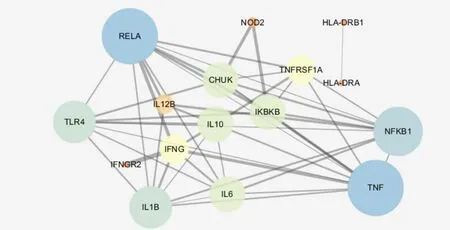
Figure 2 Interaction network of 16 genes

Table 3 Functions of 16 genes
Discussion
UC is a disease with a very complex etiology and pathogenesis, resulting in no clinically effective drugs to cure the disease. The pathogenesis of UC involves multiple pathways, so we should find out the key target genes and regulate the signaling pathways involved in this way, so as to effectively prevent and treat disease.
In this study, TTD, Drugbank, and DisGeNET databases were used to identify 315 genes related to UC. The KEGG pathway enrichment analysis of these genes was performed using the DAVID database.Among the 20 pathways with the most significant enrichment, the NF-kappa B signaling pathway includes 19 genes and 8 genes with a frequency of ≥ 9,including NFKB1 (16), RELA (16), TNF (16), IL1B(15), TLR4 (11), TNFRSF1A (10), CHUK (9), IKBKB(9). Studies have shown that under normal conditions,NF-κB is usually combined with its inhibitory protein IκB [12] and stored in the cytoplasm in an inactive form. When effective stimulation occurs, the NF-κB activation signal can activate IκB kinase (IKK). IKK can induce the phosphorylation and degradation release of IκB, so as to remove the inhibition of IκB on NF-κB, activate NF-κB, promote the transcription and expression of downstream genes, and play a regulatory role in body immunity, tissue inflammation, cell survival, proliferation, differentiation, apoptosis, etc.
Dai [13] research showed that Baitouweng Decoction plus enema and sulfasalazine retention enema can effectively alleviate the symptoms of patients with left hemicolonic UC in the acute stage.RT-PCR results showed that the expression of NF-κB mRNA in intestinal mucosal cells of the two groups of patients was significantly lower than before administration, which proved that the drug may play a therapeutic role by down-regulating the expression of NF-κB mRNA and inhibiting the release of pro-inflammatory factors. Zhao [14] research showed that mRNA and protein expression levels of NF-κB p65, IκB-α, and TLR4 in colon tissue of UC rats were significantly increased. Banxia Xiexin Decoction can down-regulate the expression of NF-κB p65, IκB-α,TLR4 mRNA and protein in the NF-κB signaling pathway of intestinal tissues, and exert its therapeutic effect on UC. The specific molecular mechanism is shown in Figure 3.
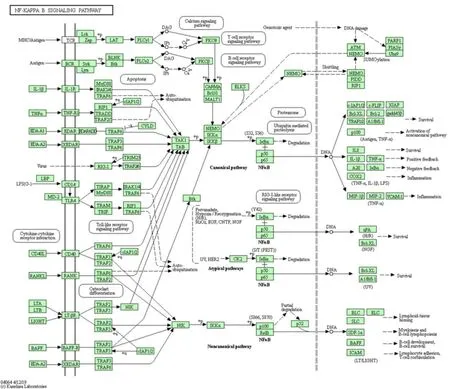
Figure 3 NF-kappa B signaling pathway
JAK/STAT signaling pathway is the main signal transduction mechanism of a wide range of growth factors and cytokines. It is not only involved in inflammatory responses, but also closely related to oxidative stress, cell damage, and apoptosis. The Jak-STAT signalling pathway involves 18 genes and 5 genes with frequency of ≥ 9, including IL6 (16), IFNG(14), IL12B (13), IFNGR2 (12), and IL10 (10). Zhao et al. [15] found that the expression of JAK2 and STAT6 protein in the colon mucosa of colitis mice treated with astragalus polysaccharide was significantly decreased,while the expression of SOCS1 and SOCS3 was significantly increased. It is speculated that astragalus polysaccharide treatment of UC may be achieved by inhibiting JAK/STAT signal activation. Niu et al. [16]found that the expression of STAT3 in mucosa of patients with UC increased and activation increased,and the expression and activation gradually increased with the increase of endoscopic lesions. STAT3 protein may be involved in the pathogenesis of UC through the IL6/JAK/STAT3 pathway. Zhao et al. [17] found that Qingchang Huashi Recipe can inhibit the activation of JAK2 and STAT3 in HT-29 cells induced by LPS and TNF-α. Qingchang Huashi Recipe inhibits the development and persistence of colon inflammation by reducing IL-6 expression, inhibiting JAK2 and STAT3 activation. It can be seen that the JAK/STAT signaling pathway plays an important role in the inflammatory disorders of UC, and JAK inhibitors can be used as a new strategy for treating UC. The specific molecular mechanism is shown in Figure 4.
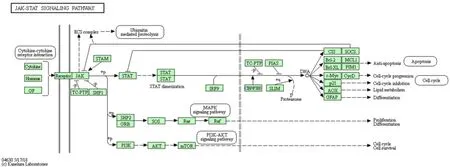
Figure 4 Jak-STAT signaling pathway
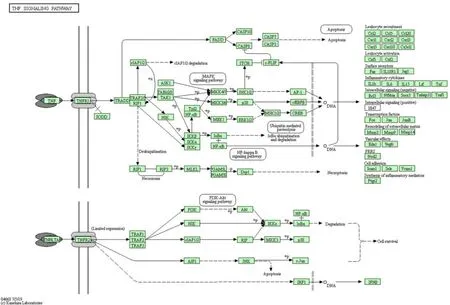
Figure 5 TNF signaling pathway
The TNF signaling pathway enriches 18 target genes from 87 genes, including ICAM1, IL18R1, TNF,PTGS2, RELA, MMP9, NFKB1, VCAM1,TNFRSF1A, CASP3, NOD2, CCL20, RIPK1, IL1B,IKBKB, TNFAIP3, and CHUK. The TNF family is an important target group for UC drugs. TNF-α belongs to the TNF family. It has a wide range of biological effects. In addition to being able to regulate the body's immune function, it is also an inflammatory factor. It combines with specific receptors on the cell membrane to achieve biological effects such as promoting cell growth, differentiation, apoptosis and inducing inflammation. Serum levels of TNF-α are related to the clinical activity of UC and Crohn's disease [18]. In addition, TNF-α can activate the three signal pathways of Caspase protease, JNK, and the transcription factor NF-kB, achieving its biological functions such as cytotoxicity, antiviral, immune regulation, and apoptosis [19]. The specific molecular mechanism is shown in Figure 5.
Twenty pathways with the most significant enrichment were analyzed. A total of 87 non-redundant genes were enriched on these pathways, and 16 genes appeared on 20 pathways with frequencies of 8 or more, including TNF, RELA, NFKB1, IL1B, TLR4,IKBKB, IL10, IL6, CHUK, TNFRSF1A, IFNG, IL12B,NOD2, HLA-DRA, HLA-DRB1, IFNGR2. We made a network diagram of the interaction between 87 and 16 UC related genes through the String database. We found that RELA, TNF, IL1B, NFKB1, IL6, and IL10 are higher in the two network diagrams, indicating that they are closely related to UC. Studies have now found that many common immune responses in UC are mediated by inflammatory cytokines, but the exact pathogenic mechanism of these small peptide molecules is still unclear. It is believed that maintaining the balance of pro-inflammatory and anti-inflammatory cytokines in the intestinal mucosa is very important for maintaining the intestinal ecological balance [20]. Excessive production of pro-inflammatory cytokines such as TNF-α, IL-6,IL-23 and inadequate production of anti-inflammatory factors such as IL-4, IL-10, IL-22, etc., leading to environmental disorders in the intestinal tract is the cause of UC Important link [21]. IL-6 is an immunoregulatory cytokine that activates the cell surface signaling complex in the inflammatory response. In the pathogenesis of UC and the subsequent formation of UC-related colon cancer tumors, IL-6 plays an important role mainly through the STAT3 signaling pathway [22].
In this study, we explored target genes that are closely related to UC through various aspects such as pathways, gene functions, and gene interactions.Finally, we found that RELA, TNF, IL1B, NFKB1, IL6,and IL10 are closely related to UC. These 6 genes may be important target genes for our new drug development.
