
冰水-溶液-炸药中耦合氢键的受激协同弛豫
2020-03-11 02:55李佶彪黄勇力孙长庆
原子与分子物理学报 2020年6期
李佶彪,黄勇力,张 希,姚 闯,张 蕾,孙长庆
(1.长江师范学院材料科学与工程学院,重庆 408100;2.湘潭大学材料科学与工程学院,湘潭 411105;3.深圳大学纳米表面科学与工程研究所,深圳 518060;4.中物院高性能数值模拟软件中心,北京 100088;5.南洋理工大学电力与电子工程学院,新加坡 639798;6.吉林大学材料科学与工程学院,长春130022 )
1 背景动机
氢键无处不在,从液态到固态,从无机到有机,从食品到药物,从细胞到生命,它的重要性固然不必赘述,但是对于氢键本质的认识以及它的定义仍在争议中不断更新[1-4].根据国际纯粹和应用化学联合会(IUPAC)2011 年推荐的定义,X-H...Y-Z中的H...Y吸引作用为二体氢键[5].其中X和Y具有比H和Z较高的电负性而带负电,譬如,碳、氮、氧、氟,等.通常称X和Y分别为质子的施体和受体.其中“ -”代表成键,即两体之间存在电荷交换或轨道交叠,而“…”代表Y的负电荷,包括Y的孤对电子或Y-Z的π键电子.
二体氢键可以发生由X-H...Y-Z向X...HY-Z以太赫兹频率的质子转移[6,7].它的作用包含静电吸引,库仑色散(极化场与其诱导的偶极子),以及在质子转移过程中所显示的部分共价特性.在特定条件下,X-H...Y显示协同性,即X-H变短会提高它对红外光的散射截面以及振动频率并伴随H...Y的伸长.这一权威定义已经被业界广泛采用并且其内容也在不断地丰富[3,4,8].遗憾的是,H...Y氢键对描述水的结构和破解冰水外场作用下所显示的反常物性具有很大的局限性.
另外,在研究分子晶体或分子集合时,人们习惯于把每个分子作为基本的结构单元考察各单元之间的相互作用以及每个分子在时间和空间域的行为.当前流行的处理方法是将分子动力学理论计算和超快时间分辨声子谱学结合探究分子的转动和平动方式、输运特性以及在某特定配位环境位置的停留时间和结构弛豫等[9,10].毋庸置疑,分子、质子和电子的输运方式是很重要的一个方面[11-15],但是,如何获得譬如尺度和能量等改变诸如密度、相变温度和热扩散系数等物理性质的因变信息的难度更大.对分子晶体而言,我们不仅需要考虑分子间的相互作用而且需要考虑分子内与分子间的耦合,以完整准确地描述氢键网络的结构,以及在时、空、能量域的受激响应和行为及其决定的物理化学性能[16].
2 耦合氢键
在1992 -2003 年间本文作者之一在采用超低能电子衍射和扫描隧道显微方法处理铜表面氧吸附时曾考虑了类氢键(O:Cu -O)的耦合作用并证明以Cu2O为基元在(110)面形成O2-:CuP:O2-单链和以Cu3O2为基元在(100)面形成耦合双链[17].上标p 表示偶极子.以Cu 取代H的耦合O2-:Cu+-O2-类氢键在反应过程中显示了其分段长度的协同弛豫特性.表面原子低配位效应导致Cu -O间距从标准的0.185 nm收缩12%到0.163 nm,而O:Cu则伸长至0.194 nm.2010 年在着手解决水的结构和冰水反常物理性质时,正式启用了水的氢键(O:H-O)耦合振子对和分段比热的概念[16].它的物理基础是通过近邻两氧的孤对电子“:”间的库仑排斥和极化,耦合原本被认为是独立的分子间的O:H非键和分子内的H-O极性共价键.2014 年在处理水合反应时进一步拓展了耦合氢键的受激协同弛豫并引入了H↔H反氢键和O:⇔:O超氢键的概念[18].2019年提出并验证了耦合氢键的拉伸张力和超氢键或反氢键斥力的组合不仅稳定氮基炸药的晶体结构,而且通过共价键收缩而储能[19-21].图1 插图所示为以H为坐标原点的耦合O:H-O键.在标准大气压和4℃温度下,它的分段长度、能量、振动频率分别为(0.1 nm,0.2 eV,200 cm-1)L和(0.17 nm,4.0 eV,3200 cm-1)H.下标L和H分别代表O:H和H-O分段.水的耦合氢键服从下列规则[16]:
1.水的四面体配位结构和氢键构型.由于氧的sp3电子轨道杂化,每个H2O分子具有电量为δ≈0.6 -0.7 e的两个质子和两对孤对电子.δ值随外场作用发生变化.为方便讨论起见,我们取δ为一个单位.在10-11-1012Pa压强和100-103K温度范围内,氧的sp3电子轨道依然保持,所以质子和孤对电子的数目守恒,耦合氢键的构型和取向守恒[22-24].即使在极高压力和温度下发生2H2O↔H3O+:HO超离子化转变时,每四个O:HO键中仅有一个反转取向而已[23,24].
2.分子转动和质子隧穿.水分子的转动和质子在两氧之间非对称位置的随机隧穿[6,7]受到严格的能量限制.在一个中心和顶角分别被水分子占据的H2O:4H2O四面体中,由于氢键取向守恒,绕中心水分子的三重对称轴转动大于60°时将会产生一个具有排斥作用的H↔H或O:⇔:O,此即破坏水的结构稳定性.譬如,单个H2O分子在二维冰面内转动120°会导致整体结构长程失序(称为Bjerrum缺陷)[25,26].另外,质子隧穿发生前必须克服氧的束缚并逃逸,而汽态水分子的H-O键(5.1eV)裂解需要在121.6 nm波长的激光辐照下才能实现[27].
3.能量交换与能量耗散.氢键网络通过HO的伸缩与外界交换能量,H-O缩短吸收并储存能量,而H-O伸长则反之.网络通过O:H热涨落或分子蒸发所耗散的能量仅限于0.2 eV范围.H-O的结合能主导冰水氢键网络的高比热值.相对H-O的4.0 -5.1 eV结合能而言,通过每条O:H非键热涨落而耗散的能量仅是H-O键能的5%而已.
4.耦合氢键的分段受激协同弛豫与极化.O:H-O键可以近似为非对称、超短程、强耦合的振子对.由于O-O的静电排斥耦合作用,外场作用使O:H-O键两端的氧沿着它们的连线以不同的长度进行相同方向的位移.O:H左端的氧的位移量总是大于H-O右端的氧,|ΔdL|>|ΔdH|,且伴随除升温外的孤对电子极化.如图1所示,无论在何条件之下,O-O间距dOO的受激变化总是通过耦合氢键的一段伸长和另一段缩短实现.O:H-O键长度的协同弛豫服从如下普遍规律[16],其中ρ表示密度:

5.氢键结合能与分段振动频率.O:H段的范德瓦尔斯势、H-O段交换作用势、以及O-O排斥耦合主导耦合氢键的三体短程作用势.而伦敦色散极化、核量子效应、自旋-自旋和自旋-轨道耦合、以及长程作用等作为平均背景.O:H-O势函数的谐振近似以及通过拉格朗日-拉普拉斯变换求解的分段振动频率表述为[28,29]:
式中kx和kC分别为x段振子和库仑排斥耦合力常数,为各自势函数在平衡点处的曲率.振子的振动位移和约化质量表示为ux和μx.Ex是分段的结合能.通过计算和实验可以直接得到分段长度和振动频率的受激弛豫,变换求解可获得分段的力常数和结合能,从而获取耦合氢键的受激弛豫的动态势能演变路径.对势函数的简谐与非线性处理的效果无明显差异,因为受激弛豫发生在近平衡点处.势函数非线性项的引入仅造成原有谱峰的微小移动而不产生任何代表新键的谱峰[30].在外场作用下,体系由原来平衡点不断地向新的平衡点弛豫.所以动态势能路径的意义较其静态更为真实、明显、丰富.
6.耦合氢键的比热与相变温度和密度震荡.根据爱因斯坦的理论,决定体系德拜比热的德拜温度正比于它的特征振动频率;此外,德拜比热的全温区积分对应体系的结合能.将德拜比热和爱因斯坦关系拓展到分段耦合氢键的情形,可以得到如下关系:

式中TVx是x分段的热裂解温度.对于O:H段,TVL=373 K是标准大气压下水的蒸发温度.根据爱因斯坦关系以及已知水的192 K的德拜温度和O:H的~200 cm-1振动频率,特征频率在3200 cm-1的H-O键的德拜温度约在3100 K.据此条件得到图2a所示的O:H-O分段比热曲线.与图2b的密度-温度震荡曲线相比发现,比热曲线的特征点对应于标准气压下汽态,液态,固态的各相边界.两条比热曲线的交点给出了在液态和固态之间、具有热缩冷胀特征的准固态(QS).准固态的相边界为密度极值点,分别对应冰点258 K和靠近熔点273 K的277 K[31].在密度-温度曲线上,273 K处并没有显示任何相变特征.那么,是否可以考虑277 K为熔化温度呢?值得注意的是,外场作用可以通过改变氢键的分段长度、能量、和振动频率以及极化特性以调节比热曲线,从而调制相应的相变温度以及氢键网络的热力学行为.
3 协同弛豫
3.1 分段长度
图1 所示为采用COMPASS 力场方法[36]计算获得的氢键协同弛豫.在压力场[16],液态(零度以上)和准固态(零度以下)温度场[16],以及(H2O)N≤6团簇分子配位场[16]作用下,耦合氢键的分段长度以“主-从”方式发生协同弛豫[34].图中箭头靠近弛豫的主动分段并指向外场变化的方向.O—O间距的任何变化总是通过其中一段伸长和另一段缩短实现,而且O:H的变化大于H-O的变化量.两段协同弛豫总是以相反的斜率和曲率进行,与外场的性质无关.电极化与低配位具有相同的弛豫特征.
3.2 振动频率
拉曼散射或红外透射谱是检测单键刚度的最有效方法.通过傅里叶变换,声子谱峰集合了具有相同振动频率或力常数的振子而与它们所处的空间位置或取向无关.物理场的扰动并不制造新的化学键.除极端条件外,物理场的作用只能改变声子的丰度(谱峰积分)、刚度(频率)、序度涨落(半高峰宽)而不产生新的谱峰.通过差分声子谱(DPS)可以过滤振子因外场作用强度变化引起的声子特征改变.差分声子谱是将两个在不同场强作用下所采集的谱进行对应谱峰面积归一化后相减.谱峰面积归一化的目的是消除实验误差.差分声子谱谱线的横轴上下分别对应声子丰度由参考基准(场强为零)向受激弛豫状态的转移.

图1 耦合O:H-O键构型(插图)及在(a)压力场[32],(b)液态冷却,(c)准固态冷却[31],和(d)(H2O)N低配位分子团簇[33]中的受激协同弛豫.箭头靠近驱动分段并指向变化方向.由于O-O的排斥耦合,O:H部分的变化相比于H-O分段总是斜率和曲率相反、变化量更大[34].静电极化与分子低配位效果相同[35].Fig.1 O:H-O cooperative relaxation under stimulus of(a)mechanical compression[32],(b)Liquid cooling,(c)QS cooling[31],and(d)(H2O)Ncluster molecular undercoordination[33].Arrows denote the master segments and point to their relaxation directions.The O:H always relaxes more than the H-O in opposite slopes and curvatures,irrespective of the stimulus applied or the structural phase because of the persistence of O-O repulsive coupling[34].Electrification has the same effect of molecular undercoordination,resulting in the supersolidity[35].
图3 所示O:H-O键在温场、力场、配位场作用下的声子频率的协同弛豫.声子弛豫测量结果与图1 所示的长度弛豫计算结果相对应.H-O和O:H段的振动频率在外场作用下发生反向频移.分段振动频率的蓝移与它的长度收缩对应一致.压致H-O声子红移至3100 cm-1.离子水合层内的H-O键和O:H非键的特征频率为3500和70 cm-1.离子静电极化与分子低配位作用相同.值的关注的是,液态水和冰的表皮共享以H-O振动频率为3450 cm-1为特征的超固态.各谱线的谱谷对应参考基准的特征频率.体相冰和水的H-O振动频率分别为3150 和3200 cm-1.
3.3 动态势场

图2 (a)O:H-O分段比热曲线决定(b)冰水在常压下的相结构和密度变化[31].具有冷胀热缩且相边界可调特征的准固态的相边界对应着密度极值,并且分别接近冰点和熔点(258,277)K.分段比热的比值ηL/ηH决定密度随温度变化的斜率.比热值低的分段服从常规的热胀冷缩规则而另一段则反之.外场作用通过改变分段的结合能和振动频率调制各相变温度.大量低配位分子降低1.4 nm水滴的冰点到205 K[38].Fig.2 O:H-O segmental specific heat derived structure phase and mass density variation[31].Subjecting to cooling expansion,the QS phase boundaries correspond to the extreme densities and close the(TN,Tm)for bulk at(258,277)K.The segmental having a lower specific heat follows the regular rule of thermal expansion while the other segment does it contrastingly.The QS boundary is retractable by external field.The 1.4 nm droplet has the least density and freezes at 205 K[38].
耦合氢键的核心在于它的动态作用势[28,29].作用势的平衡位置对应键的长度和能量.在外场驱动下,势的平衡位置从初始平衡状态向另一稳态过渡,也即分段长度和能量发生弛豫.在真实情况下,只有通过改变外场作用的强度才能探测到物质性质的变化.但是,通过谱学和衍射实验无法直接测定势场[42-44].只能通过解耦和还原进行正则变换,解析拉格朗日-拉普拉斯耦合振子对力学,才可以将直接测量的分段长度和振动频率转换为相应的力常数和结合能,从而得到如图4所示的动态势能曲线.这样,我们获得了耦合氢键在力、热、电、以及配位场作用下分段长度(图1)、振动频率(图3)、能量(图4)的协同弛豫的完整图像和定量信息.这些物理基本参量加之价电子的行为决定冰水的本质.
4 冰水范例
4.1 密度震荡
对于冰水,人们不禁要问为什么冰的密度比水的低?关于这个问题的讨论始于1611 年.伽利略和哥伦布在意大利的佛罗伦萨分别从质量密度、物体形状和表面张力等方面持续了几天的争论无果[45].2013 年,20 多名来自世界各地的学者再聚此地研讨了一个星期,以纪念伽利略和哥伦布讨论400 周年.仍其说不一,至今尚无定论[46].典型的观点认为过冷水是由高密度的四配位和低密度的低配位链状分子的畴状组合.降温提升以畴分辨的低密度相的比例,结果导致浮冰现象的发生[38,47].
比较图2 的分段比热和冰水密度震荡,不难发现如下对应关系,

耦合氢键的分段比热差异和温致协同弛豫的建立给出了截然不同的物理图像[31].对比图2 的比热和密度随温度变化的行为可见,决定冰水密度变化的是耦合氢键分段比热的比值,dρ/dt∝ηL/ηH-1.在任一温度都有两个比热值,比热数值低的分段服从常规的热胀冷缩定律,而另一段由于O-O排斥作用反之.密度ρ1/3∝dL+dH且随O-O间距增大而降低.在液态和固态第I相,d ρ/dt∝ηL/ηH-1 <0,密度随温度升高而降低.O:H段主导热胀冷缩且其变化量大于H-O分段.结果导致表观与常规一致但是机理截然不同的热胀冷缩现象.在低温第XI相,d ρ/dt∝ηL/ηH-1≈0.由于德拜比热近零值,两段长度和能量对温度不敏感,声子频率基本恒定[48].由于∠O:H-O键角从167°冷扩张至173°,密度微弱降低[31].
现在来关注在权威相图中有待标注的准固态.在准固态中,d ρ/dt∝ηL/ηH-1 >0,H-O键主导O:H-O的温致弛豫.H-O的冷缩量小于由其驱动的O:H的冷膨胀,结果导致在准固态相中O-O间距受冷增大、密度降低.在分段长度变化的过程中,∠O:H-O键角从160°扩张至167°,也增大O-O的间距.所以,浮冰只发生在准固态.准固态的密度从4℃时的最大值1.0 降低至-15℃时的0.92 g/cm3.在第I相,密度因降温回升至0.94 g/cm3.可见,由耦合氢键分段比热主导的长度弛豫唯一地决定准固态和各相冰水密度的变化速率.
4.2 冰水表皮

图3 水在(a,b)受热[39],(c)受压[40],以及(d)25℃水和-(15 -20)℃冰表皮[34]的差分声子谱.(a,b)O:H和H-O声子频率显示协同性.冰水表皮共享以H-O振动频率3450 cm-1为特征的超固态,并决定了(d)插图所示的冰表皮的超滑和水表皮超韧的特性[41].Fig.3 DPS profiles for water under(a,b)heating[39],(b)compression[40],and the skins of 25 ℃water and -(15 -20)℃ice[34].Water and ice share the same supersolid skin characterized by the identical H-Obond stiffness of 3450 cm-1,which responsible for the shows the slipperiness of ice and the skin toughness of liquid water(inset d)[41].
液态水的表皮具有在自然界中最高的韧性[41]而冰的表皮具有最低的摩擦系数[49,50].人们认为冰的表皮覆盖一层水而水的表皮有一层冰[51,52].类液态表皮不仅作为摩擦润滑剂而且是连接两块冰的粘接剂.但是冰-冰的摩擦系数可以高达0.58.人们通常采用摩擦生热[53]、压致复冰-熔点降低[52,54]、纳米流变[55]、低配位分子滚动[56]、以及低配位导致的超固态-低频声子高弹性和极化静电排斥解释冰润滑和水皮高韧性行为[16,57].
由图1d和图3d 可见,分子低配位导致HO收缩刚化、O:H非键伸长弱化,并伴随电子极化.因此,体相水和表皮水的H-O键能分别为4.0 和4.6 eV[16].X-射线光电子发射谱揭示,体相、表皮和汽相水的氧1s能级分别为536.6、538.1、539.7 eV[58,59].氧1s能级的深移标志H-O键长缩短、键能增强[60].液态微射流紫外光电子谱测量结果表明,入射到水的深层和表层电子的束缚能分别为3.2 eV和1.6 eV.当水滴尺度减小到只含5 个分子时,入射到液态水滴中的电子束缚能从1.2 降低至0.4 eV[61-64].所以,X-射线和紫外光电子发射谱直接证明了H-O收缩导致的O 1s能级钉扎深移以及非键电子极化.
所以,冰的表皮不是水,水的表皮也不是冰,而是冰水表皮共享以3450 cm-1振动频率的H-O为特征的超固态[16].正是表皮的超固态决定如图3d插图所示冰的超滑和水表皮的超韧特性.冰-冰的高摩擦系数源于接触界面两侧O:H软声子的共振耦合以及复冰效应(冰块因表皮超固态粘连).超固态起因于O:H-O协同弛豫和相应的强极化效应——独具低密度(标准值的3/4)、高弹性、电排斥、高热扩散系数、低比热、高熔点、低冰点、疏水等特性.也正是超固态决定了纳米液滴在微通道输运过程中显示的超流行为以及纳米气泡和水滴的过冷和过热现象以及它们的化学活性和力学、热学稳定性.
4.3 电场极化
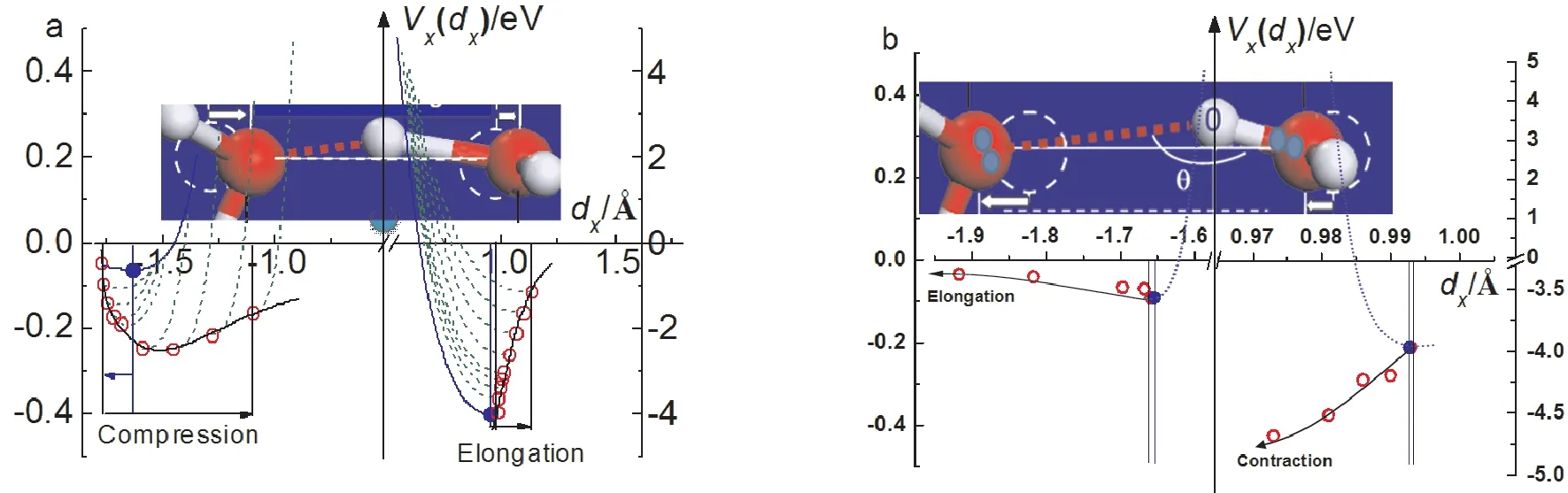
图4 耦合氢键在(a)受压(由左至右:5,10,…,60 GPa)和(b)低配位拉伸作用下((H2O)N团簇(从右到左:N=6,5,…,2)的势能动态协同变化曲线[28,29].氢键两端的氧沿相同方向位移且O:H的变化量总是大于H-O的变化.任一段长度的缩短伴随势阱变深.蓝点对应无O—O排斥作用时的平衡点,红色空心圆对应于在不同外场强度作用和不同配位数条件下的平衡位置.Fig.4 Potential paths for the(a)mechanically compressed(l to r:5,10,60 GPa)and(b)undercoordination stretched O:H -O bond((H2O)N,r to l:N=6,5,…,2)[28,29].Both Odislocate in the same direction by different amounts and the O:H relaxed more than the H-O(note the H-O scales in both axis).The blue dots correspond to initial equilibrium without O—O repulsion and the red open circles are locations under different strengths of the external fields.
通过电荷与杂质注入、带电衬底、电荷流动、疏水限域、直流电场、等离子体放电等极化手段进行液态水的深加工来进行制氢、水提纯、和相变温度调制是缓解环境和能源危机、改善人类生存条件的重要手段.历史上著名的电场对水的极化现象包括水桥、相变温度调制和电致蒸发.阿姆斯壮(Armstrong)[65]在1893 年发现在室温和105V/m电场强度下,在两个盛满水的水杯之间可以形成稳定的、韧性极高的、厘米长度的水桥.杜佛(Dufour)在1861 年发现可以通过施加电场调节水的结冰温度[66].浅川(Asakawa)在1976 年发现水的电致快速蒸发[67].
电场具有定向极化分子和通过振动频率调节相变温度的双重作用.电场的极化作用与分子低配位作用效果相同.与外加电场平行的表面张力增大,而在垂直电场方向的表面张力则降低.沿电场方向的H-O键缩短、O:H非键弱化.以图5a和b所示的通过水合盐分解的离子对氢键极化的差分声子谱为例,离子通过其径向极化缩短和刚化它的水合层内H-O键而伸长弱化O:H非键,形成具有高熔点、低冰点以及高结构序度和韧性的超固态.量子计算结果显示电场极化拉伸O—O间距,提高水分子的偶极矩[68,69].通过极化可以改变氢键两段的强度而调制相变温度[70-73].
所以,电场极化导致的超固态决定了阿姆斯壮(Armstrong[65])水桥的温度和力学稳定性.垂直电场方向表面张力的弱化、O:H的弱化、沸点的降低,显著地提升了Asakawa电致蒸发效率(浅川效应).通过施加电场改变结冰和熔化温度在生物活细胞的低温保存中具有重要价值[35].
所以,耦合氢键唯一地确定了冰水的结构和性能.液态水是由超固态表皮包裹的、具有四面体结构的静态均匀单相而动态强涨落的单晶.耦合氢键分段的受激协同弛豫、极化和分段比热差异主导冰水在受激过程中所呈现的超常自适应、自愈合、高敏感、和强记忆等性能.表1 列出了氢键的分段长度和振动频率的受激弛豫特征以及相应的冰水属性.表面张力与电子极化程度正相关.熔点温度Tm正比于H-O键能EH,冰点TN和沸点TV温度正比于O:H结合能EL.电场作用与分子低配位的作用效果相同[35].
值得注意的是,液态水升温通过缩短H-O键而储能,降温则反之.这一行为提供了热水结冰快的能量转换的前提[75,76].冰水受压致H-O弱化伸长而O:H刚化缩短.相应的能量改变决定了复冰现象-压制熔点降低、冰点升高[77-79].分子配位数降低或通过水滴纳米化提高低配位分子数目比例,可以提升液滴的熔点并降低其沸点和冰点.除共享极化效应外,分子低配位效应与压强作用对氢键长度弛豫的效果相反.表2 列举了在外场作用下冰水显示的典型反常物性以及从耦合氢键受激弛豫和极化角度的理解.
(2)检查范围与方法:对子宫病灶范围、边界、回声、血流等情况予以观察,详细记录病灶与周边超声图像;取微泡悬浮液0.8ml经患者肘部浅静脉团注,期间观察造影剂灌注过程,灌注顺序依次为子宫浆膜层、子宫肌层、子宫内膜层。
5 水合反应
水合反应的本质是通过溶解可溶物质向溶剂中弥散注入不同电量、不同几何形状和尺度的电荷以调制溶液的氢键网络和物理化学性质[18].可注入电荷包括离子、电子、质子、孤对电子、偶极分子,等.除质子以H3O+和孤对电子以HO-形式与溶剂分子作用外,其它注入电荷并不与水分子分享或交换电荷或产生新的化学键.溶质的作用只是通过O:H-O的弛豫、H↔H反氢键和O::O超氢键的产生、屏蔽极化、负离子间的排斥、和溶质内低键序或溶质分子低配位导致的键收缩改变氢键网络的结构和能量分布.
如图5 插图所示,离子和电子以偏心填隙式形成(±):4H2O:6H2O结构提供一个被其近邻偶极水分子屏蔽的电场而极化其最近邻4H2O和次近邻6H2O,导致水合层内H-O键收缩、声子频率蓝移.体积较小的阳离子完全被其水合层屏蔽,所以它们的水合层体积不受其它离子存在或溶质浓度的影响.而体积较大的阴离子不能被它们的水合层完全屏蔽,显示长程作用.所以,溶质浓度的升高增强近邻阴离子间的排斥作用,其水合层体积也逐渐变小且自身电场变弱.这种效果随阴离子半径增大和电负性降低会愈加明显,服从霍夫梅斯特盐溶液溶解蛋白质能力的序列[91].

表1 O:H-O分段长度和振动频率的受激协同弛豫以及由电子极化决定的表面张力γ.Tm∝EH,TN∝TV∝EL.参考标准:dL0=1.6946 Å,dH0=1.0004 Å,ωH0=3200 cm-1,ωL0=200 cm-1,ΘDH=3200 K,ΘDL=192 K.Table 1 O:H-O segmental cooperative relaxation in length,vibration frequency,and surface stress with respect to dL0=1.6946 Å,dH0=1.0004 Å,ωH0=3200 cm-1,ωL0=200 cm-1,ΘDH=3200 K,ΘDL=192 K up on excitation by heating,compression,molecular undercoordination(skin,cluster,droplet,nanobubble).(Note:ΔΘDx∝Δωx;ΔTm∝ΔEH;ΔTN∝ΔTN∝ΔEL;EDL(electric-double-layer))

表2 典型的有关冰水溶液的反常物理化学性质的耦合氢键解析Table 2 Interpretation of the typical anomalies of water and ice.

图5 (a,b)NaCl,(b)HCl和(d)NaHO溶液的差分声子谱.(b)中插图所示离子的偏心填隙式占位[92],替位式占据的酸根/碱基与其近邻水分子间形成的(c)H↔H反氢键和(d)O:⇔:O超氢键[18].Fig.5 DPS profiles for(a,b)NaCl(c)HCl and(d)NaHO solutions within set(b)showing the eccentric interstitial occupancy(c)H↔H anti-HB and(d)O:⇔:O super-HB formation between the central(H3O+;HO-)with its one of the neighboring H2O molecules[18].
路易斯酸碱[93]的水合引入了额外的质子和孤对电子而打破水的质子和孤对电子数目守恒[18].如图5c和d插图所示,注入的质子和孤对电子分别形成以H3O+和HO-为中心替位的(H3O+;HO-):4H2O结构单元并将单胞内一条氢键转换为具有排斥功能的H↔H反氢键和O:⇔:O超氢键.这种排斥使溶剂的H-O键伸长并弱化而呈现低于3100 cm-1的差谱特征.按库仑定律,电荷之间的作用力与两电荷的乘积正相关.所以,在间距相同的条件下,超氢键的两对孤对电子的排斥是反氢键两个质子的四倍之多.与图3c所示的水受压声子谱相比,H↔H反氢键和O:⇔:O超氢键的排斥作用远高于纯水在室温条件下受压结冰所需的1.33 GPa压强的效果.这种排斥呈现长程衰减的特征.
图5c酸溶液的在3650 cm-1位置的谱谷表明阴离子优先占据酸溶液的表面,提供较强的局域电场并通过极化缩短H-O悬键而使其频率从3610 cm-1移至3650 cm-1,同时高密度阴离子的极化也屏蔽悬键声子振动的检测信号.图5d碱溶液中阳离子的极化信号由于其短程效应而被淹没.在3610 cm-1处的尖峰源于HO-因低键序导致其H-O单键的收缩刚化.所以,HO-的H-O单键与水表面的H-O悬键相同.类似的,双氧水分子的水合也引入一对孤对电子,所以双氧水溶液和溶质的H-O声子谱显示与碱水的谱学类似的特征.溶质的H-O键特征峰呈现在3550 cm-1,溶剂的H-O键的受压弱化也呈现低于3100 cm-1的谱峰.
双氧水和碱溶液中的O:⇔:O超氢键和乙醇溶液中的H↔H反氢键对近邻水分子的排斥导致溶剂H-O键伸长和弱化而释放能量,其效果是溶解过程中溶液自升温.如图6 所示,0.1 摩尔浓度的碱溶液和乙醇溶液温度分别可达55℃和32℃;双氧水溶液在0.16 摩尔浓度时升高到30℃.原理上,酸和盐水合离子的极化导致的HO收缩刚化吸受热量,但是酸和盐溶质在反应过程断键则释放能量.所以在溶解过程中,它们的溶液并没有显示明显的温度变化.
有机分子如醇、醛、酚、酸等的外围均附有不同数目的悬键质子和孤对电子.这些极性溶质分子与近邻水分子间作用通过各自质子和孤对电子的组合形成不同数目的O:H吸引,H↔H或O:⇔:O排斥,以改变氢键网络和溶液的物理化学性质.所以,在有机溶质的水溶液中,除溶质偶极分子与水分子相互作用外,在溶质-溶剂界面还存在O:H-O氢键、H↔H反氢键和O:⇔:O超氢键作用.

图6 不同摩尔浓度的(a)氢氧化(锂,钠,钾),(b)双氧水和(c)乙醇(插图)在水合反应过程中的溶液自升温测量结果.Fig.6 Self-heating of the molar-ratio-concentrated(a)(Li,Na,K)OH,(b)H2O2and(c)Ethanol(inset)solutions.
6 氮基炸药
拓展耦合氢键、反氢键和超氢键的概念到由碳、氮、氢、氧构成的含能分子体系,有助于从分子内和分子间耦合作用的角度理解含能分子系统的结构稳定性和分子内强键的能量存储机制.类比冰水溶液,含能分子体系通过共价键的收缩储能.分子间因相互排斥和吸引作用的平衡使结构稳定,而分子间结合强度决定炸药的感度[21].
6.1 全氮五唑
图7a和b所示为全氮五唑阴离子范例的优化结构和受力分析[19,94].实验证明,全氮N5-阴离子只有在酸性环境中才能稳定存在[95-97].环中相邻N原子共价成键,它们的外层电子轨道发生sp2杂化,孤对电子占据第三条轨道(图7b).N的第7个价电子(2s2p5)的集合与一个外来电子形成π键芳香结构.所以氮环内存在N-N共价键和N:⇔:N超氢键以及双芳香π键.在N5-:(4H3O+或2H3O++3H4N+)复合体系中,外围酸根间H↔H反氢键排斥压缩它们的H-O或H-N键.酸根间斥力之和径向拉伸N:H-O/N耦合氢键导致H-O/N收缩、N:H伸长,而弱化内环的N:⇔:N排斥.环内N-N共价键因N:⇔:N弱化而缩短.量子计算结果显示,由于酸根的介入,N-N键从1.38 Å收缩到1.32 Å而N:HN的N:H长度扩展到2.10 Å.N:H-O键的N:H的长度在2.17 -2.26 Å范围,均长于水在4 ℃时的标准O:H参考长度1.70 Å.所以,在这个体系中所有的共价键都因其收缩而储能.整个五唑阴离子复合体系的稳定性取决于N:H的拉伸和外环H↔H排斥的组合.如果N:H因受冲击断裂,整个系统将崩溃、爆炸发生.
6.2 硝基炸药
如图7c所示,一个TATB(C6H6N6O6)分子以C6苯环为中心,每个碳原子与一个氮原子连接成键;每隔位N原子分别与两个O原子和两个质子相连接.C6环芳香结构的大π键决定体系的层间作用.每个N和O都经受sp3轨道杂化而分别产生一对和两对孤对电子.这些孤对电子和与N相连的质子是分子体系的基本功能单元.除每个分子的6 个沿边缘方向具有吸引作用的O:H非键外,近邻分子间通过它们各自的质子和孤对电子形成O:H非键吸引、H↔H或O:⇔:O排斥而稳定体系.由于每个分子的质子数目少于孤对电子的数目,所以分子间只有O:⇔:O和O:HC键存在,正是前者的排斥和后者的拉伸作用力稳定分子体系的结构.另外,耦合氢键的拉伸缩短它的共价键分段,超氢键的排斥同样压缩与它直接相连的N-O共价键.所以体系通过共价键的缩短而储能.通过测量分子晶体的变温和变压的声子谱可以证明耦合氢键的存在.具有排斥作用的反氢键和超氢键的存在只能改变氢键和现有成键的长度、能量和振动频率,而它们本身并不提供新的成键.

图7 典型含能分子晶体的基元结构和作用力示意图.(a,b)五唑氮环通过N:H-O/N与外围H3O+或NH4+的拉伸作用弱化N-N的排斥使N-N和H-O/N缩短.外环酸根间的排斥合力拉伸N:H-O/N键[19,94].(c)TATB分子通过它的30对孤对电子和6 个质子以及芳香π键与近邻分子作用形成耦合N-H:O/N拉伸氢键和O:⇔:O排斥超氢键.Fig.7 Molecular motifs for the(a)cyclo-N5-:4H3O+with the force diagram,and(c)the cyclo-N5-:(3H3O++2NH4+)of bond relaxation.For(c)TATB,the black spheres are carbon,green ones are H,red ones are oxygen and the blue ones are N.Each N carries one and Otwo pairs of lone pairs.At the critical concentration of stabilization,the radial N:H-Otension by the circumferential H↔H repulsion not only stabilizes the cyclo-N5-complexes but also stores excessive bond energy.The configuration shortens all the covalent bonds and lengthens the N:H nonbond,see context for discussion.reprinted with permission from[19,94].
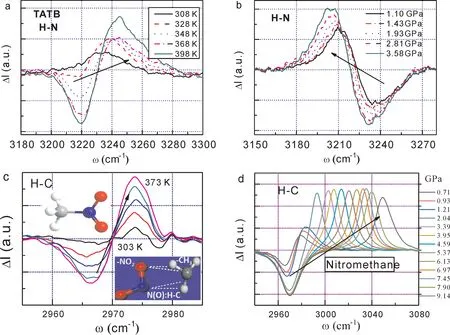
图8 TATB炸药中H-N键的反常(a)热致收缩刚化和(b)压致膨胀弱化以及硝基甲烷中H-C键的(c)热致和(d)压致刚化[20].(c)插图所示为硝基甲烷的分子结构和分子间耦合氢键的形成.Fig.8 H-N bond and H-C bond(a,c)thermal stiffening and(b,d)compressive softening for the(a,b)O:H-N bond in TATB and(c,d)the O:H-C bond in nitromethane assemblies[20].
图8 所示的TATB和硝基甲烷(CH3NO2)的变温变压声子差谱证明O:H-N/C耦合氢键确实存在.但因O—N和O—C的耦合强度以及H-N和H-C的电负性差的区别,测量结果有明显的差异.O:H-N的H-N键显示与水的O:H-O中H-O键完全相同的热致收缩刚化和压致膨胀弱化的特征(见图3).相对O:H-N而言,O:H-C的H-C键仅显示热致收缩刚化而没有压致伸长弱化.X-Y分子间的排斥耦合作用力满足fO—O>fO—N>fN—N>fC—O>fC—N的顺序.如表3所列,H-O/N/C的受激频移确有差异.TATB的C-NO2和N-O键以及CH3NO2的C-N和N-O键服从常规的热胀压缩的规律,而TATB的CNH2与硝基甲烷中的C-H键显示热致和压致收缩刚化.这些键对温度和压力的响应方式与它们是否在耦合作用范围之内以及耦合强度有关.H-N/C和C-NH2的反常行为证明了含能分子体系确实存在超氢键排斥和受拉伸的耦合氢键,而且它们的组合不仅稳定分子间的平衡而且通过成键的收缩而储能.耦合氢键的非键强度决定炸药的感度,它的受激断裂引起爆炸.

表3 水、TATB、硝基甲烷中成键的温致和压致刚度弛豫.括号内所示数据为振动频率的改变.Table 3 Mechanical and thermal response of the intramolecular O:H-O/N/C bonds for water,TATB and nitromethane assemblies.
7 总结展望
作为新的尝试,耦合氢键、反氢键、超氢键的提出和验证使我们不仅能够定量破解水的结构和多项关于冰水的谜题而且加深了对酸碱盐和有机溶液的氢键网络和属性以及氮基炸药的结构稳定性和储能机理等的系统认知:
1)水是由单一耦合氢键组成的、由超固态表皮包裹的、最简单的、静态高度有序而动态涨落的分子晶体.它在温场、力场、电场和配位场作用下的分段长度、能量和振动频率的协同弛豫和极化以及分段比热差异决定它的超常自适应、自愈合、高敏感、热稳定等性能.分段比热不仅决定了具有热缩冷胀的准固态相而且划分了常压下的各相边界.外场作用通过氢键协同弛豫和爱因斯坦关系调节相变温度.分子低配位和电极化具有相同效果导致具有高弹、疏水、润滑、低比热、高稳定性的超固态;电场的方向性极化和对相变温度的改变决定超固态水桥,电致结冰和蒸发;压力场与低配位对氢键弛豫的效果相反;除升温外,氢键受激弛豫导致电子极化.长度、能量和价电子的能量和空间与行为是决定可测物理量变化的关键.
2)水合反应以电子、质子、离子、孤对电子、偶极子的方式弥撒注入电荷并通过耦合氢键协同弛豫、H↔H反氢键、O:⇔:O超氢键、静电屏蔽极化、溶质键收缩以及溶质间的相互作用调制溶液的氢键网络和性能.酸碱水合提供质子和孤对电子并以酸根和碱基的方式存在并替位式占据(H3O+;HO_):4H2O的中心,分别将每个单胞内的一条氢键转换成反氢键和超氢键;盐水合分解的离子和注入电子以偏心间隙式形成(±):4H2O:6H2O结构,提供屏蔽电场极化近邻水分子而形成超固态水合层而非形成新的化学键;有机溶质水合同样造成界面氢键弛豫并形成反氢键和超氢键.水合过程中,阴离子间的排斥弱化离子的局域电场和水合层体积;低键序溶质的键收缩造成声子蓝移.
3)反氢键或超氢键的排斥压力与耦合氢键的拉伸张力不仅稳定炸药分子间的平衡而且通过共价键的收缩使其储能.前者主导炸药感度,后者决定储能密度.炸药中的耦合氢键协同弛豫与X:H-Y中的X-Y排斥耦合强度正相关.
4)仅在X-Y的排斥足够强而且H-Y的电负性差足够大的条件下耦合氢键才能发生协同弛豫.此即决定了耦合氢键的适用范围——并非所有的X:H-Y都可以发生受激协同弛豫.O:H-O是极端理想的情形.X和Y包括所有电负性大于氢的元素而且H也可以被电负性较低的金属譬如Cu原子取代.作为氢键的基本要素,孤对电子和质子或正离子应该得到足够的关注和重视.
作为有机分子和生物分子的基本功能单元,孤对电子和悬键质子是实现DNA、蛋白、细胞、药物、食品、到生命体的功能以及信号加工和传递的基石.分子间通过各自的质子和孤对电子与近邻分子通过形成耦合氢键、反氢键、超氢键相互作用.所以,考虑分子间与分子内作用的耦合,拓展耦合氢键、反氢键、超氢键和电子极化的概念到分子电子动力学具有深远的意义.
致 谢:感谢吉林大学王志刚教授的有益讨论和湘潭大学唐志旭同学的实验帮助.
猜你喜欢
汽车实用技术(2022年5期)2022-04-02
波谱学杂志(2021年3期)2021-09-07
今日农业(2021年7期)2021-07-28
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
舰船科学技术(2021年12期)2021-03-29
沈阳师范大学学报(自然科学版)(2019年2期)2019-06-19
当代陕西(2019年6期)2019-04-17
国外科技新书评介(2016年12期)2017-04-17
天津师范大学学报(自然科学版)(2016年4期)2016-12-14
中学化学(2015年12期)2016-01-19
