
离心条件对丙酮沉淀法提取尿蛋白质的影响
2020-02-10 06:40:48周冬冬
基础医学与临床 2020年2期
李 京,周冬冬,孙 伟
(中国医学科学院基础医学研究所 北京协和医学院基础学院 药理系,北京 100005)
近年来,尿液(urine)逐渐成为寻找疾病标志物的重要来源[1],但是,尿蛋白的浓度相对较低。因此,尿蛋白的提取方法的优化非常重要,此外,健康人尿蛋白质组的个体差异分析可以为蛋白质组研究提供稳定性评价,排除疾病标志物发现过程中个体差异的影响,为蛋白质组学研究提供背景,具有重要的临床价值[2]。
在蛋白提纯过程中,目前已有的报道中丙酮沉淀方法提取尿蛋白所用离心转速不尽相同[3],在不同离心条件下,离心温度是否对蛋白提取有影响也没有相
应报道。本项研究比较了不同的离心条件下(转速和温度)尿蛋白提取效率和质谱分析结果,从而确定最佳尿蛋白离心方法。另外,以往个体差异分析的样品数目相对较少,尿蛋白质组的研究深度不足[4],本研究拟对36例健康人(18例男性和18例女性)的尿液蛋白质组的个体差异进行分析,为后续的差异蛋白质组学和靶向蛋白质组学分析提供依据。
1 材料与方法
1.1 材料
试剂:二硫苏糖醇(dithiothreitol, DTT)、碘乙酰胺(iodoacetamide IAM)、血管紧张素Ⅱ(Sigma-Aldrich公司);胰蛋白酶(质谱级)(Promega 公司);乙腈、甲酸、H氟乙酸、碳酸氢铵均为色谱级(Merk公司);氯化钠、乙醇和盐酸(北京化学试剂公司);4×十二烷基硫酸钠聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate polyacrylamide gel electrophoresis, SDS-PAGE)上样缓冲液、NuPage预制胶和蛋白标准品(Invitrogen公司); SDS(USB公司)。
1.2 方法
1.2.1 尿液样品: 应用于尿液离心方法优化的样本为来自10名健康男性和10名健康女性的晨尿,每人留取24 mL,等量混合后,以3 000 r/min离心30 min,去掉尿沉渣。应用于个体差异尿液蛋白质组学分析的样品来自18名健康男性和18名健康女性[年龄为(44±14)岁],每个样品取用2 mL处理,并用3 000 r/min离心30 min,去掉尿沉渣。
1.2.2 尿蛋白提取及酶切: 在尿蛋白离心条件优化过程中,每个样品加1 mol/L DTT至终浓度0.02 mol/L,95 ℃加热5 min,冷却至室温后加入1 mol/L的IAM至终浓度0.05 mol/L,室温避光45 min。加入6倍体积的-20 ℃预冷丙酮混匀后,放置在-20 ℃半小时。之后将尿液样品按表1方法进行处理,离心10 min后去上清,将4组沉淀吹干后加入 0.02 mol/L Tris溶解。应用Bradford方法进行蛋白浓度的测定,并取等量蛋白进行SDS-PAGE凝胶电泳。4种方法各取100 μg蛋白进行蛋白质样品酶切。
在非标记定量的尿液蛋白质组学分析的样本处理中:离心条件选择20 ℃,12 000×g,其他操作步骤同上。
1.2.3 质谱分析: 尿蛋白离心条件优化的质谱分析方法:多肽样品溶解于0.1%甲酸水溶液,经毛细管色
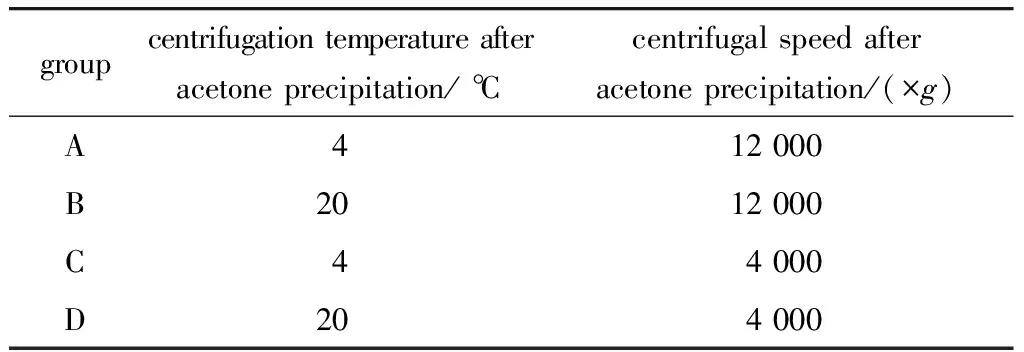
表1 4种不同的离心处理方法Table 1 Four different centrifugal methods
谱柱(100 cm ×75 μm,3 μm)进行分离,多肽的洗脱液梯度为5%~30% B液(0.1%甲酸,99.9%乙腈),流速为600 nL/min, 洗脱时间为60 min。洗脱的肽段用质谱仪分析鉴定,分析模式采用数据依赖采集模式,参数如下: 一级全扫描范围为350 m/z ~1 550 m/z, 分辨率为12万, 二级扫描碰撞能量为30%, 分辨率为3万, 采集方法为高速度模式, 电荷筛选范围(+2 ~+7),动态排除时间为30 s,最大离子注入时间0.045 s。
非标记定量质谱分析方法:样品的液相分析条件同上,质谱采集方法采用数据非依赖模式,使用30个可变窗口采集,一级扫描分辨率为12万,质荷比范围为400 m/z~900 m/z,二级质谱扫描分辨率为3万,碰撞能量为32%,AGC为1e6,最大离子注入时间为0.05 s。
1.3 统计学分析
1.3.1 定性分析: 质谱数据应用Proteome Discovery软件进行数据库检索,数据库为SwissProt人类蛋白质组学数据库(20 430条序列,2018年)。其他参数为:胰蛋白酶酶切,误切位点为2个,母离子和子离子的质量精确度为10 ppm和0.02 ku,固定修饰为半胱氨酸的脲基甲基化(+58.00 ku),可变修饰为甲硫氨酸氧化(+16.00 ku),蛋白质N末端乙酰化(+0.98 ku),蛋白质K端甲酰化(+43.01 ku)。
1.3.2 定量分析: 应用Progenesis QIP (版本 4.1, 美国) 进行定量分析,将其电荷数筛选为+2到+5,导出的质谱数据,使用Mascot软件 (Matrix Science公司,版本2.5.01)进行数据库检索,检索参数同上。检索结果导入Progenesis QIP后,对相应蛋白和多肽进行定量分析。
1.3.3 非标记定量分析:使用Spectronaut Pulsar软件分析,Spectronaut将保留时间、质核比等与数据库中比对,对蛋白质进行鉴定和定量分析。
1.3.4 最小样本量的计算:使用R语言包(pwr.t. test)计算最小样本量[5],公式如下:n=ceiling[pwr.t.test(d=(fold-1)/cv,sig.level=alpha,power=1-b, alternative=“two. sided”)$n]。
2 结果
2.1 不同离心方法提取尿蛋白
在相同离心转速的条件下,4 ℃温度下得到的蛋白量均高于20 ℃温度下得到的蛋白量;在相同离心温度条件下,高离心转速12 000×g得到的蛋白量高于低离心转速4 000×g得到的蛋白量,在4种离心条件中,A组方法(4 ℃,12 000×g)得到的蛋白量最多(表2)。SDS-PAGE分离结果(图1A)显示,不同离心方法得到的尿蛋白样品条带相似,条带明显清晰,并未有蛋白质降解条带,实验重复性良好。
2.2 不同离心方法得到人体尿蛋白的质谱鉴定
定性重复性平均为90.26%±1.43%,定量重复性为97.08%±0.25%。说明各种方法的实验重复性良好,实验结果可靠(表2)。
相同转速条件下,4 ℃条件下鉴定的蛋白数、多肽数、谱图数和可定量的蛋白数目比20 ℃方法多。相同温度条件下,高转速(12 000×g)离心鉴定的蛋白数、多肽数、谱图数和可定量蛋白数比低转速(4 000×g)多,4种方法中,4 ℃,12 000×g离心条件下的鉴定结果最好。
定性蛋白中,有617个蛋白是4组中均鉴定到的(图1B),说明了4种方法的可靠性,其中只在A组鉴定到的蛋白有84个,明显高于其他组(14~26个),说明在A组(4 ℃,12 000×g)的条件下提取到的蛋白种类更多。
在定性蛋白中A 组和B组差异有统计学意义(P<0.05);在定量蛋白中A组和C组差异有统计学意义(P<0.05);在肽段中A组和C组差异有统计学意义(P<0.05)。

A.SDS-PAGE for four different centrifugation methods; B. comparison of the urine protein identified by four centrifugation methods图1 4种离心方法的结果比较Fig 1 Comparisons of four centrifugation methods results

表2 不同离心方法处理人尿蛋白的质谱鉴定结果Table 2 LC/MS/MS result of human urinary proteome using different centrifugation n=8)
*P<0.05 compared with group A.
2.3 不同离心方法鉴定蛋白的属性比较
4种方法下得到的尿蛋白的等电点测定结果显示,在等电点6~7范围内的蛋白质数是最多的,等电点大于9的蛋白数是最少的,4种方法鉴定到的蛋白质等电点的分布没有差异。A组(4 ℃,12 000×g)鉴定的蛋白等电点分布在偏酸性和中性者多于其他组(图2A)。
4种方法得到的尿蛋白分子量测定结果显示,分子质量为30~40 ku的蛋白数目最多,分子质量为140~150 ku蛋白最少,4种方法鉴定的蛋白分子质量分布范围无差异。在A组(4 ℃,12 000×g),低分子质量的蛋白略多于其他组(图2B)。
4种方法误切率的比较结果显示,定性分析中,各组并没有明显差别,在定量分析中,A组的误切率较高,与B、C组差异有统计学意义(P<0.05)(图2C)。
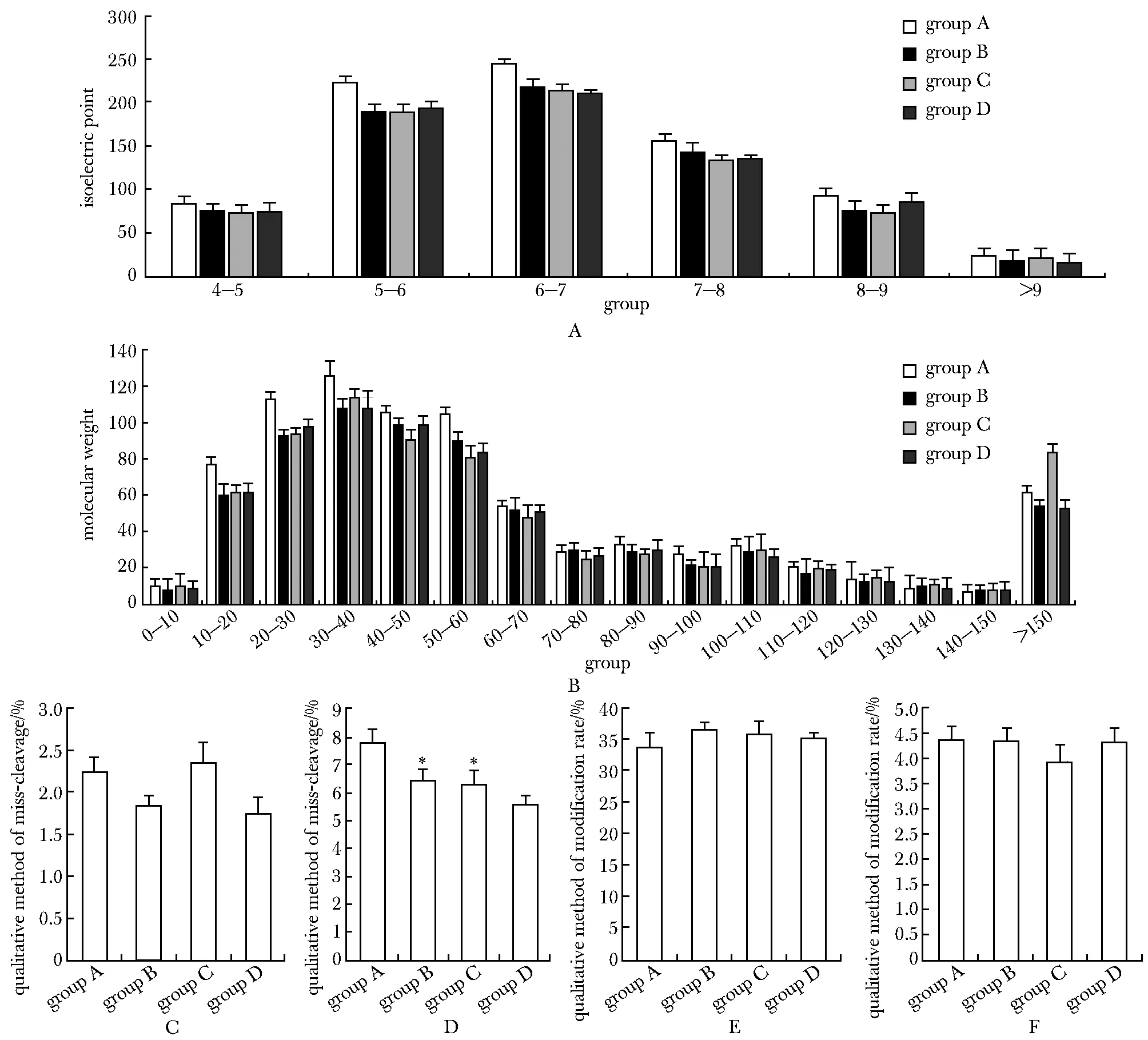
A.the isoelectric point distribution of urine protein by four centrifugation methods; B.the molecular weight distribution of urine protein by four centrifugation methods; C.the qualitative method of miss-cleavage of urine protein by four centrifugation methods; D.the quantitative method of miss-cleavage of urine protein by four centrifugation methods; E.the qualitative method of modification rate of urine protein by four centrifugation methods; F.the quantitate method of modification rate of urine protein by four centrifugation methods; the post-translational modification ratio of urine protein by four centrifugation methods
图2 4种离心方法鉴定蛋白的等电点和分子质量分布情况以及胰蛋白酶误切率和翻译后修饰的比较
在蛋白质翻译后修饰比较中,4种可变修饰所占比例在4种方法中无明显差别。表明不同离心方法对翻译后修饰并无明显影响(图2D)。
2.4 健康人尿蛋白质组个体差异分析
36个样本的混合样本建立的非标记定量数据库中,共鉴定出3 032个尿蛋白。尿液样品非标记定量分析结果显示,定量到的蛋白个数为2 300。去除技术重复性大于50%的蛋白后,结果显示, 每个样本平均定量的蛋白数为1 379个。尿蛋白质组的个体间差异中位数为0.635, 男性和女性个体间差异的中位数分别为0.628/0.476(图3A)。
2.5 个体差异与技术差异分析
尿蛋白的技术差异中位数为0.211,男性/女性的中位数CV为0.218/0.222,个体差异和技术差异相关性分析(图3B)显示,92.5%的尿蛋白个体差异CV值大于技术差异CV值。
2.6 差异蛋白质组学所需最小样本量计算
当倍数变化为2时,尿液样品的平均最小样本量约为15个。男性和女性尿液样本的平均最小样本量为15个和11个(表3)。

A.inter-individual variation of urinary proteomes; B.the correlation of technical variation and individual variation图3 尿蛋白质组的个体差异分析Fig 3 Inter-individual variation of urinary n=8)
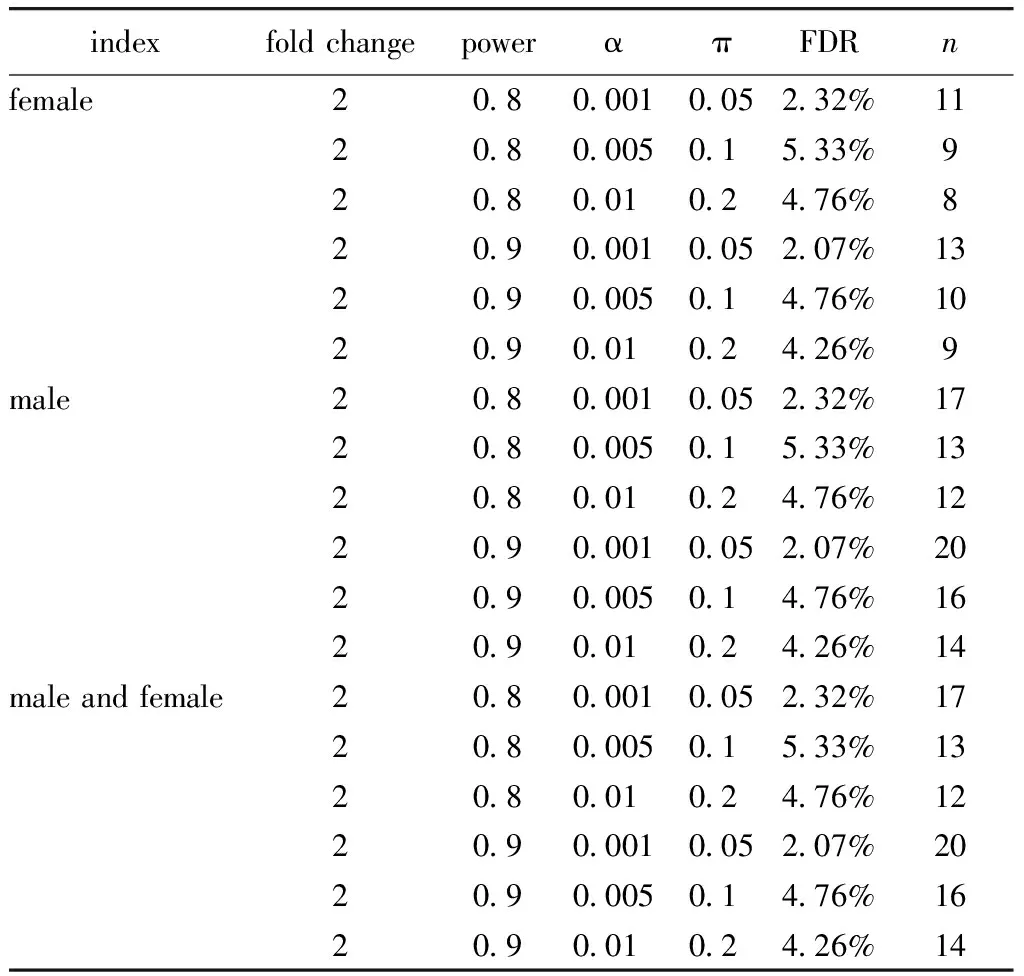
表3 基于尿液非标记定量方法的最小样本量评估
power was the power of the statistical test, α was the significance level, and π referred to the estimated proportion of truly deferentially expressed proteins among all of the identified proteins; FDR referred to the false discovery rate.
3 讨论
本次研究有以下几个方面可以进一步探讨。
在离心温度方面,以往文献报道,蛋白在溶液中的溶解度主要受到温度和蛋白大小的影响[6],蛋白质在溶液中,水化层可以保护蛋白质与蛋白质之间的相互作用。破坏水化层会导致蛋白质沉淀[7],以往研究表明,低温条件会促进水化层的破坏,同时低温下丙酮可以更好地保持蛋白的活性[8]。因此,低温(4 ℃)离心条件下得到的蛋白更多。
在离心转速方面,以往文献发现使用丙酮沉淀方法不足以沉淀分子质量较小的蛋白。但是当转速高时会促进小分子质量蛋白质的沉淀[9],因此高转速可以提高小分子质量蛋白的回收。
在胰蛋白酶的误切率方面,根据研究结果推测A组误切率高主要是由于鉴定小分子质量的蛋白肽段导致的,具体的机制需要进一步探究。
之前很多项研究使用不同的蛋白定量方法进行个体差异分析,如非标记定量法[10]、同位素标记相对和绝对定量技术[11]。非标记定量方法是基于峰强度和一级质谱信号进行定量。但是,一级质谱数据的准确性和特异性较差,具有相同质核比的杂质峰可能会错误地计入目标肽中。因此,这种方法可能会放大真实变化[12]。本文采用的数据非依赖的定量方法和之前研究中的同位素标记相对和绝对定量技术的方法是基于二级质谱信号定量,比一级质谱定量更准确,但是在同位素标记相对和绝对定量技术方法中,杂质的干扰可能会压缩信号的变化导致结果不准确[13],而数据非依赖的定量方法定量不存在这一缺点,定量更加准确。
据以往文献报道,血浆蛋白质组学的个体差异的中位数为60.3%[14],和本研究所鉴定到的健康人尿液个体差异63.5%相近,说明尿液蛋白质组的稳定性与血浆蛋白质组类似,可以应用于大规模的临床样品分析。
总之,本研究确定了丙酮沉淀提取尿蛋白的最佳离心方法(4 ℃,12 000×g),并且利用这种方法和非标记定量方法评估了尿蛋白质组的个体差异以及最小样本量,为尿蛋白提取提供了最优离心方案,也为尿蛋白组的大规模临床应用奠定了基础。
猜你喜欢
军事文摘(2022年16期)2022-08-24 01:50:52
中国典型病例大全(2022年11期)2022-05-13 17:54:50
基础医学与临床(2022年1期)2022-01-21 05:22:06
国际口腔医学杂志(2019年3期)2019-05-31 10:09:26
时代英语·高一(2018年4期)2018-09-14 10:53:14
天然产物研究与开发(2018年2期)2018-04-04 02:01:12
妈妈宝宝(2017年4期)2017-02-25 07:01:26
中国现代药物应用(2016年12期)2016-03-06 11:54:25
医学研究杂志(2015年11期)2015-06-10 06:44:03
现代检验医学杂志(2015年2期)2015-02-06 02:01:08
