
基于能量传递可调色度ZnNb2O6: Dy3+, Eu3+荧光粉的制备及其发光性能研究
2020-01-10 02:35梁柏鑫易双萍胡耕樵方志雄赵韦人
广东工业大学学报 2020年1期
梁柏鑫,易双萍,胡耕樵,方志雄,赵韦人
(广东工业大学 物理与光电工程学院,广东 广州 510006)
目前,荧光粉广泛应用于发光二极管(Light Emitting Diode, LED)中,通过LED中紫外芯片泵浦红绿蓝(RGB)三色荧光粉,合成白光[1]。随着芯片制造技 术的快速发展,芯片的发光波长已经扩展到近紫外甚至能量更高的紫外区域,为RGB三基色系统提供了更广的研究空间。然而,三基色荧光粉之间的重吸收、荧光寿命不一致等,导致样品发光效率降低。另外,红色荧光粉光输出效率明显不足[2],导致在可见光光谱的红色成分较弱,色温有所偏差。而通过掺杂稀土或过渡金属离子的无机材料,由于其良好的化学和热稳定性和多颜色发光特性而得到广泛研究[3-5]。学术界近年来致力于通过掺杂稀土离子进而改善三基色荧光粉的上述问题。
含有NbO6八面体结构的铌酸盐由于其非线性光学、铁电、压电、光电和光催化等诸多优异性能而引起了人们广泛的关注。大多数铌酸盐具有较宽的宽带发射、较高发光效率以及良好的化学稳定性等[1,6]。在紫外光激发下,GdNbO4、La3NbO7、CaNb2O6和Ca2Nb2O7在300~600 nm具有较强的带宽[7-8],证明铌酸盐具有较宽的电荷迁移带[9-11],有利于光吸收,使其具有较大的潜在应用价值,如M2+Nb2O6(M=Ni, Co,Fe, Mn, Zn, Mg, Ca, Cd)系列的铌酸盐也已经被广泛研究报道。其中,铌酸锌(ZnNb2O6, ZN)是人们熟知的铌酸盐材料之一。研究表明,掺杂或无掺杂ZnNb2O6均可作为优异的激光发光材料[12-13]。Wang Rui等[14]报道了在980 nm激光激发下,上转换ZnNb2O6: Ho3+/Yb3+中存在基质与稀土离子的能量传递现象,最终增强发射了具有较强绿色和红色的发射带。Guo Jing等[15]发现,在365 nm激发下共掺杂Al3+离子可以改善ZnNb2O6: Dy3+的发光强度,具有较大的可调稀土离子掺杂浓度。
由于Dy3+可同时发射蓝、黄两色光,Eu3+可发射红色光,Dy3+/Eu3+共掺杂可有望实现白光发射,但其在ZnNb2O6基质中掺杂还未见报道。鉴于此,本文采用高温固相法制备了ZnNb2O6:xDy3+,yEu3+(0≤x≤0.15,0≤y≤0.32)新型荧光粉,系统地研究了ZnNb2O6的光致发光特性。从CIE色度坐标图可以得出,在ZnNb2O6: 0.08Dy3+,yEu3+样品中,随着Eu3+离子浓度的增加,色度可调至白光区域;其次,确认了Dy3+/Eu3+共掺杂ZnNb2O6荧光粉在紫外激发下的发光特性和能量传递机制,对Dy3+离子到Eu3+离子的能量传递现象及其机理进行了较为详细的探索,表明ZnNb2O6:0.08Dy3+, 0.03Eu3+是可用于紫外激发下的白光荧光粉。
1 实验步骤
1.1 样品合成
通过高温固相反应制备了ZnNb2O6:xDy3+,yEu3+(0≤x≤0.15,0≤y≤0.32)一系列的粉末样品。实验用原材料包括ZnO(99.9%,阿拉丁)、Nb2O5(99.9%,国药集团化学试剂有限公司)、Dy2O3(99.9%,麦克林)和Eu2O3(99.9%,麦克林)。按照化学计量比称量并在玛瑙研钵中混合后研磨40 min. 然后,将混合均匀的粉末样品装入氧化铝坩埚中并置于高温管式炉中煅烧,在空气气氛下加热至800 ℃,保温6 h. 最后,将所得样品在炉中冷却至室温并重新研磨成细粉,用于进一步测量研究。
1.2 表征方法
样品的物相分析采用MSAL XD-213型X射线衍射仪,仪器参数设置为在36 kV管电压和20 mA管电流下,辐射源为Cu Kα射线(λ=0.154 06 nm),以0.02°步长在10°~70°的范围内连续扫描。通过配备有(450 W,Osram)的FLS-980荧光分光光度计测量激发光谱、发射光谱和荧光寿命。样品的形貌采用SU8220型场发射扫描电子显微镜,仪器参数设置二次电子分辨率为0.8 nm和1.1 nm. 除非特殊说明,所有测试均在室温下进行。
2 结果和分析
2.1 物相分析
ZnNb2O6、ZnNb2O6: 0.08Dy3+、ZnNb2O6:0.03Eu3+和ZnNb2O6: 0.08Dy3+, 0.03Eu3+等样品的XRD衍射谱如图1所示。4种样品的衍射峰几乎与标准卡片JCPDS 76-1827相匹配,没有观察到额外的衍射峰,表明Dy3+/Eu3+或Dy3+, Eu3+共掺杂稀土离子并没有改变ZnNb2O6晶体结构。此外,根据JCPDS标准卡片,ZnNb2O6具有正交晶系,其空间群为Pbcn(60),晶格参数为a=1.420 8 nm,b=0.572 6 nm,c=0.504 nm,Z=4且体积V=0.410 0 nm3。考虑到Zn2+(0.09 nm,CN=8), Dy3+(0.119 nm, CN=8)和Eu3+(0.106 7 nm,CN=8)之间的离子半径,推测Dy3+和Eu3+可能取代在ZnNb2O6基质中的Zn或Nb格位[16]。

图1 ZnNb2O6; ZnNb2O6: 0.03Eu3+; ZnNb2O6: 0.08Dy3+; ZnNb2O6:0.08Dy3+, 0.03Eu3+和标准卡片的XRD衍射图Fig.1 XRD patterns of some representative samples ZnNb2O6;ZnNb2O6: 0.03Eu3+; ZnNb2O6: 0.08Dy3+; ZnNb2O6: 0.08Dy3+,0.03Eu3+ and the standard XRD data of ZnNb2O6 (JCPDS 76-1827)

图2 各样品的扫描电镜图Fig.2 SEM micrographs of samples
图2显示ZnNb2O6, ZnNb2O6: 0.03Eu3+, ZnNb2O6:0.08Dy3+和ZnNb2O6: 0.08Dy3+, 0.03Eu3+等样品的扫描电镜图(SEM)。可以注意到,所获得的粉末颗粒大小在几微米之间,颗粒的形貌和大小通常在高温固相反应和充分研磨之后形成的。从图2(a)可以看出,不同形态的颗粒自由组合形成具有粗糙表面的颗粒。图2(b)和图2(c)是在不同放大倍数下掺杂了不同稀土离子的ZnNb2O6: Dy3+/Eu3+荧光粉。与图2(a)~图2(c)相比,图2(d)中样品的形态发生了较明显的变化,在共掺杂离子之后,形成具有光滑表面和不规则形状的块状颗粒。
2.2 光谱分析
图3为ZnNb2O6基质的激发和发射光谱图。在456 nm监测波长下,样品在220~320 nm的激发光谱范围内具有较宽电荷迁移带。同时观察到在267 nm波长的激发下,ZnNb2O6基质以456 nm为中心的蓝色发射峰,而在614 nm处的发射峰则是由于铌酸盐基团产生的缺陷或杂质所引起的[17]。在ZnNb2O6的正交晶系结构中,Zn、Nb原子与6个O2–阴离子配位,分别形成ZnO6和NbO6八面体。Pullar R C等[18]还报道了八面体之间通过公共的边或角相互连接,并且在bc平面上形成二维可拉伸的薄片形状. 由于俘获电子的重组,八面体配位的[NbO6]7–基团是蓝光区域发射范围的有效发光中心。

图3 ZnNb2O6的激发和发射光谱图Fig.3 Excitation and emission spectra of ZnNb2O6
图4(a)为在614 nm的监测波长下ZnNb2O6:0.04Eu3+荧光粉的激发光谱图。图中显示以394 nm为中心的窄带激发峰和在350~500 nm波长范围内的多个激发峰主要是由于Eu3+离子的f-f能带跃迁引起的,分别对应于7F0→5D4(363 nm),7F0→5L6(394 nm),7F0→5D3(416 nm)和7F0→5D2(468 nm)。图4(b)显示了在394 nm波长激发下,样品ZnNb2O6:xEu3+(x=0.01,0.02, 0.04, 0.06,0.08)的发射光谱图,位于576,592,612和654 nm处的几个发射峰,它们分别对应于Eu3+离子的5D0−7FJ(J=0,1,2,3)跃迁[19-20]。Eu3+的发射强度随着Eu3+浓度的增加而增强,可以观察到掺杂浓度为0.04时达到最大值。

图4 ZnNb2O6: xEu3+的激发和发射光谱图Fig.4 Excitation and emission spectra of ZnNb2O6: xEu3+
图5显示了在576 nm的监测波长下,ZnNb2O6:0.08Dy3+的激发光谱位于267 nm处的宽带激发峰和在350~500 nm的一系列窄带激发峰。将ZnNb2O6:0.08Dy3+的激发光谱与ZnNb2O6基质的激发光谱进行比较可以发现,源自于[NbO6]7–基团中的O2–-Nb5+电荷迁移的基质吸收峰比Dy3+的4f-4f 特征激发峰强。由此推断ZnNb2O6基质具有很强的吸收紫外光的能力。此外,位于267 nm处的激发峰表明在掺杂稀土离子的ZnNb2O6基质中存在从[NbO6]7–基团到稀土离子Dy3+能量传递的发生。同时由Dy3+的4f-4f跃迁可知,350~500 nm的弱激发峰分别对应于6H15/2→6P7/2(354 nm)、6H15/2→4I11/2(366 nm)、6H15/2→4I13/2(388 nm)、6H15/2→4G11/2(426 nm)、6H15/2→4I15/2(454 nm)和6H15/2→4F9/2(478 nm)[21-22]。

图5 ZnNb2O6: 0.08Dy3+的激发光谱图Fig.5 Excitation spectra of ZnNb2O6: 0.08Dy3+
在267 nm的激发波长下,ZnNb2O6:xDy3+(x=0.01,0.02, 0.04, 0.06, 0.08, 0.10和0.15)的发射光谱如图6所示,可以看出Dy3+的最佳离子掺杂浓度为0.08,样品呈现出较强的白光(CIE色度坐标为x=0.316 6,y=0.335 7)。图6中581 nm处由电偶极子跃迁4F9/2→6H13/2引起的发光强度明显高于489 nm处由磁偶极子4F9/2→6H15/2跃迁引起的发光强度,这表明Dy3+离子在基质中占据的是非对称中心。同时,样品呈现出白光是由于基质本身受激发辐射出蓝光,Dy3+的特征发射为黄光,两者以一定比例产生的。此外,以基质激发的发射峰强度高于在以Dy3+离子的特征(354 nm)激发的发射峰强度(见图7)。正如Wang Tao等[1]报道的基质发射峰的存在意味着从基质到Dy3+的能量传递是不完全的。正因如此,从基质到Dy3+的能量传递在可调色度和有效紫外激发白光中起关键作用。

图6 ZnNb2O6: xDy3+的发射光谱图Fig.6 Emission spectra of ZnNb2O6: xDy3+

图7 ZnNb2O6: 0.08Dy3+在基质激发和Dy3+的特征激发下的发射光谱图Fig.7 Comparative emission spectra of ZnNb2O6: 0.08Dy3+ under host and Dy3+ characteristic excitations
图8显示在581 nm的监测波长下ZnNb2O6:0.08Dy3+, 0.25Eu3+的激发光谱。图中267 nm处的带宽峰为基质的吸收带宽。插图中350~500 nm的激发峰主要由于Dy3+和Eu3+的特征跃迁引起。在267 nm激发下,ZnNb2O6: 0.08Dy3+,yEu3+(y=0.03~0.32)发射光谱如图9所示,图9中的插图表示Eu3+的浓度与样品发光强度之间的关系。可以发现,Eu3+的发光强度随着Dy3+的发光强度降低而增强,Eu3+掺杂浓度为0.25时达到最大值。Dy3+的4F9/2能级(约21.5×103cm–1)高于Eu3+的5D0能级(约19.1×103cm–1),通过以声子辅助的非辐射弛豫方式,从Dy3+到Eu3+离子的传递了部分能量[23]。此外,能量传递与Eu3+离子晶体场的局部对称性密切相关。据文献报道,Eu3+离子处于低对称位置时,Eu3+的5D0→7F2电偶极子跃迁与Eu3+的5D0→7F1奇偶性允许磁偶极跃迁相比占得一定优势[24]。于是,从5D0→7F2跃迁强度高于5D0→7F1跃迁,证明Eu3+离子同样占据非对称中心晶格位置。最后,在267 nm激发下,样品接近白光(CIE色度坐标为x=0.343 1和y=0.320 0),归因于基质辐射出蓝光和Dy3+/Eu3+共掺杂的特征发射。
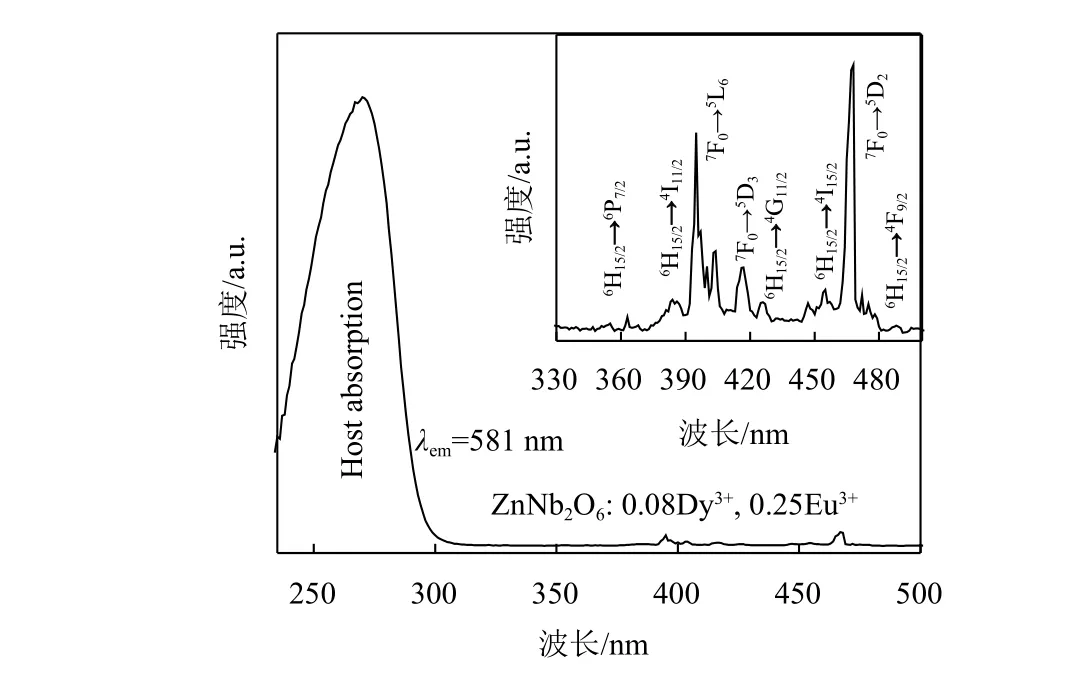
图8 ZnNb2O6: 0.08Dy3+, 0.25Eu3+的激发光谱图Fig.8 Excitation spectra of ZnNb2O6: 0.08Dy3+, 0.25Eu3+

图9 ZnNb2O6: 0.08Dy3+, yEu3+ (y=0.03~0.32)的发射光谱图 Fig.9 Emission spectra of ZnNb2O6: 0.08Dy3+, yEu3+ (y=0.03~0.32)
2.3 荧光衰减曲线
为进一步研究ZnNb2O6基质中Dy3+向Eu3+能量传递的过程,如图10所示,在354 nm激发并在581 nm监测下,测量了ZnNb2O6: 0.08Dy3+,yEu3+(y=0.03~0.32)的光致发光衰减曲线。采用二阶指数衰减模型拟合荧光衰减曲线,公式为[25]

其中A1和A2是拟合常数;I是在t时刻的发光强度;τ1,τ2代表荧光寿命。平均衰减时间τ值同样采用二阶指数拟合,公式如式(2)所示[26]。


图10 ZnNb2O6: 0.08Dy3+, yEu3+的荧光衰减曲线Fig.10 The luminescence decay curves of ZnNb2O6: 0.08Dy3+, yEu3+phosphors
对于ZnNb2O6: 0.08Dy3+,yEu3+,其中y=0, 0.03,0.05,0.07,0.10,0.15,0.20,0.25和0.32. 平均衰减时间τ值分别对应0.200 3,0.181 0,0.150 0,0.139 4,0.106 4,0.093 3,0.074 6,0.047 7和0.028 6 ms。可以观察到,当Dy3+的掺杂浓度固定在0.08,随着Eu3+离子浓度的增加,样品的平均寿命τ值降低。因此,由ZnNb2O6:0.08Dy3+,yEu3+中Dy3+寿命的逐步下降,进一步证实了共掺杂稀土离子ZnNb2O6基质中存在Dy3+向Eu3+的能量传递。
2.4 色度坐标图
图11为ZnNb2O6,ZnNb2O6:xDy3+,ZnNb2O6:xEu3+和ZnNb2O6: 0.08Dy3+,yEu3+的色度坐标图,交叉点表示CIE色度坐标位置。首先,ZnNb2O6基质的色度坐标落在蓝光区域,色度坐标值为(0.146 7,0.089 1)。其次,ZnNb2O6:xDy3+荧光粉的发光颜色从蓝光区域跨到黄光区域,随着Dy3+离子浓度的增加,相应坐标从(0.231 0, 0.262 2)变化至(0.343 9, 0.359 2)。这表明合成单掺杂Dy3+离子的ZnNb2O6基质接近标准白光(0.33, 0.33)。还注意到ZnNb2O6:xEu3+的坐标主要落在红光区域上,并且相应的坐标值从(0.564 2, 0.425 3)变化至(0.623 6, 0.371 6)。最后,随着ZnNb2O6:0.08Dy3+,yEu3+样品中的Eu3+离子掺杂浓度的增加,色度坐标主要落在在白光区域,相应的色度坐标变化范围为(0.343 1, 0.320 0)~(0.427 6, 0.308 8)。

图11 (a) ZnNb2O6, (b) ZnNb2O6: xDy3+, (c) ZnNb2O6: xEu3+, (d)ZnNb2O6: 0.08Dy3+, yEu3+的色度坐标图Fig.11 Evolution of CIE chromaticity coordinates of (a) ZnNb2O6, (b)ZnNb2O6: xDy3+, (c) ZnNb2O6: xEu3+, (d) ZnNb2O6: 0.08Dy3+,yEu3+
2.5 能量传递机制
化合物中的敏化剂(Dy3+)与激活剂(Eu3+)发生的共振能量传递过程主要通过交换作用(Rc<0.5 nm)和电多极−电多极相互作用(Rc>0.5 nm),两者关键取决于敏化剂和激活剂之间的临界距离(Rc)。根据Blasse的理论,RDy-Eu能够表示为[27]

式中,V是晶胞的体积(V=0.410 0 nm3),N是晶胞中的分子数(N=4),x是Dy3+的发射峰强度处在无掺杂(Eu3+)时样品发射峰强度一半时的掺杂浓度。因此,临界浓度RDy-Eu计算约为0.840 nm。
非辐射能量传递主要有3种机制:辐射再吸收、交换作用和电多极相互作用。在本文中,由于Rc大于0.5 nm,表明交换作用的可能性很小。并且只有当吸收光谱和发射光谱有较大的重叠时,辐射再吸收才会占主导作用。因此,在本文中能量传递是通过电多极相互作用。根据Dexter的电多极相互作用能量传递和Reisfeld近似理论的描述,可以得到式(4)[28]。

其中η0和η代表在无掺杂和掺杂Eu3+的情况下Dy3+的发光量子效率;因子C是Dy3+和Eu3+浓度的总含量;n=6,8,10的值分别对应于电偶极矩−电偶极矩(d-d)、电偶极矩−电四极矩(d-q)和电四极矩−电四极矩(qq)相互作用。由于值η0/η不易获取,因此可通过相应衰减时间的比率(τs0/τs)近似计算[29],如式(5)所示。

其中τs0和τs是无掺杂和掺杂Eu3+的情况时敏化剂Dy3+的寿命。如图12(a)~(c)所示,曲线τs0/τs∝Cn/3揭示了在n=6,8,10处的关系。可以发现,当n=8时为最佳线性拟合,这表明从敏化剂Dy3+到激活剂Eu3+离子的能量传递机制遵循非辐射电偶极矩−电四极矩作用。

图12 Cn / 3与Eu3+的τs0/τs依赖关系Fig.12 Dependence of τs0/τs of Eu3+ on Cn / 3
图13描绘了Dy3+和Eu3+离子,以及它们在ZnNb2O6:Dy3+, Eu3+中的能量传递能级图。基质受激发吸收光子能量,触发O2–→Nb5+或者O2–→Zn2+的电荷转移,然后传递至Dy3+。此外,部分能量从Dy3+的4F9/2能级转移到Eu3+的5D0能级。受激发的Eu3+通过交叉弛豫的方式跃迁至5D0能级并辐射出荧光迁移到Eu3+的7Fj(j=1,2,3)能级,最终增强了Eu3+发红光[30]。
3 结论
在本文中,采用高温固相法合成ZnNb2O6,ZnNb2O6:xDy3+,ZnNb2O6:xEu3+和ZnNb2O6: 0.08Dy3+,yEu3+荧光粉。研究表明,在紫外光的激发下,ZnNb2O6基质发射出较强的蓝光。通过实验和理论分析,Dy3+和Eu3+离子之间的临界距离为0.840 nm。ZnNb2O6:0.08Dy3+,yEu3+中Dy3+和Eu3+的能量传递机制遵循的是非辐射电偶极矩−电四极矩作用。合成的单掺杂Dy3+离子的ZnNb2O6荧光粉接近标准白光,从ZnNb2O6: 0.08Dy3+,yEu3+荧光粉的CIE色度坐标表明,通过调节共掺杂Dy3+/Eu3+的比例可以调节发光颜色并且落在白光区域。据此所获得的荧光粉可在紫外汞灯白光源和其他固态照明技术中具有一定的潜在应用。

图13 ZnNb2O6: Dy3+, Eu3+在能级跃迁上的电荷转移和能量传递的过程Fig.13 Energy level scheme of ZnNb2O6: Dy3+, Eu3+ with electronic transitions and ET Process
猜你喜欢
延边大学学报(自然科学版)(2021年4期)2022-01-14
陶瓷学报(2021年5期)2021-11-22
中华民居(2021年4期)2021-11-18
陶瓷学报(2020年6期)2021-01-26
贵州农机化(2020年3期)2020-12-24
世界有色金属(2019年12期)2019-08-14
中国计量大学学报(2018年2期)2018-07-12
中国兽医杂志(2016年5期)2016-06-27
照明工程学报(2016年3期)2016-06-01
连环画报(2016年4期)2016-05-05
