
IGF-1 上调miR-155 表达对新生大鼠缺氧性肺动脉高压肺组织损伤的影响及其作用机制
2020-01-02 05:41于秀石魏丽丽马克涛司军强张忠双田俊杰鲁广森
中国比较医学杂志 2019年12期
于秀石,朱 佳,魏丽丽,马克涛,司军强,张忠双,3∗,田俊杰,罗 淑,鲁广森
(1.石河子大学医学院,新疆 石河子 832000; 2.新疆维吾尔自治区人民医院,乌鲁木齐 830000;3.石河子大学新疆地方与民族高发病教育部重点实验室,新疆 石河子 832000)
新生儿缺氧性肺动脉高压(hypoxia-induced pulmonary hypertension, HPH)是常见的威胁新生儿生命的急危重症,疾病早期表现为肺血管痉挛,若及时发现并进行治疗则可抑制血管痉挛,否则疾病发展到晚期则表现为肺血管重塑,发生不可逆的组织变化,此时期救治困难、死亡率高[1-2]。 目前其发病机制尚不清楚,已知血管舒缩因子的平衡失调在发病过程中起了重要作用[3-4]。 在HPH 的发病机制研究发现缺氧诱导因子1α (hypoxia-inducible factor 1α, HIF-1α)和内皮素-1(endothelin-1, ET-1)的上游调节因子,参与HPH 的发生与发展[5]。miR-155 属于小分子RNA,能通过与靶基因3’或5’非编码区(untranslated region, UTR)的结合,调控靶基因的表达,参与机体的各种生物调控过程,如miR-155 在抑郁症患者的血浆中的表达与多种炎症因子相关。 胰岛素样生长因子1(insulin-like growth factor1, IGF-1)能有效促进外周神经再生,具有非选择性神经营养作用,已有动物实验研究发现新生大鼠缺氧缺血性脑损伤后脑室内给予IGF-1,明显减轻脑损伤区的神经元缺失[6]。 在研究对HPH 的机制时,IGF-1 上调miR-155 表达的作用非常重要,但miR-155 在肺损伤中的作用鲜有报道[7];且国内尚无新生儿HPH 发生与IGF-1 相关的研究。 故而本研究通过建立HPH 新生大鼠模型,以尼莫地平为工具药,探索在HPH 发病过程中,IGF-1 的作用机制,HIF-1α 和ET-1 的生理功能,现将结果报道如下。
1 材料和方法
1.1 实验动物
清洁级Wistar 大鼠180 只,雌雄不限,日龄3 ~5 d (大鼠新生儿期),体质量(20 ± 5) g,购自海军军医大学动物中心,实验动物生产许可证号[SCXK(沪)2019-0009]。 依据应用实验动物的3R 原则给予适当关照。 饲养及无菌手术在复旦大学医学院实验动物科学部屏障动物实验设施进行[SYXK(沪)2019-0026],置于全自动调节低压舱,饲养条件:室温21℃~25℃,相对湿度50%~65%,光暗循环12 h/12 h,自由饮水摄食。 实验动物福利伦理审查号:2019012304。
1.2 主要试剂与仪器
尼莫地平(国药准字H43020645,规格:每片30 mg,湖南百草制药有限公司);鼠源性单克隆抗体(Anti-β-actin)(美国Amgen 公司);ELISA 试剂盒(美国R&D 公司);BCA 蛋白定量试剂盒(BCA protein assay kit)(美国BioVision 公司);TaqMan®MicroRNA Reverse Transcription kit 试剂盒(美国ABI 公司);Gel-Doc 凝胶成像分析系统(美国UVP公司);Leica Qwin 图像分析系统(德国Leica 公司); NanoDrop 2000 蛋白/核酸分析仪(美国ThermoFisher 公司);高速离心机(美国Beckman 公司);LightCycler480 高通量实时荧光定量PCR 仪(美国罗氏公司);TRIzol 裂解液(美国Invitrogen 公司)和RNase-free 水(美国ThermoFisher 公司)。
1.3 实验方法
1.3.1 实验分组
新生大鼠均适应性饲养1 周,随机分成3 组:模型组、给药组及空白对照组。 将模型组和给药组新生大鼠置于自制全自动调节常压低氧舱内(大气压约50 kPa,氧浓度10%),制造间断性低氧环境,以1.5 L/min 的速度输入含10%的氮氧混合气,予以氧浓度仪监测,维持氧浓度在9.5%~10.5%范围内,控制二氧化碳浓度小于0.5%。 通过放置小风扇维持低氧舱仓内气体均匀,通过减压阀和电磁阀调节舱内压力,予以无水氯化钙、钠石灰吸收箱内的水蒸气与二氧化碳。 昼/夜为12/12,每天缺氧8 h,连续2 周。 分别于缺氧2、4、8、12 d 检测肺动脉平均压(mean pulmonary arterial pressure, mPAP),mPAP≥6.5 pg/mL,且具有较强时间依赖性,则证明成功建立HPH 大鼠模型[8]。 缺氧同时给药,其中给药组每天灌胃给予3 mg/kg 尼莫地平。 空白对照组除不低氧,即大气压约50 kPa,氧浓度10%,其余条件相同,两组大鼠放于同一房间以相同饮食饲养,模型组及空白对照组每天灌胃给予生理盐水。分别于缺氧2、4、8、12 d 依照随机数字表法抽取模型组、给药组及空白对照组大鼠各10 只进行相关检测指标的监测。
1.3.2 mPAP 检测
各组大鼠在被观测时间点经氯胺酮腹腔内注射麻醉后,固定实验大鼠,消毒颈、胸部皮肤,行气管插管,给予机械通气,潮气量:2 ~3 mL/min,监测新生大鼠尾部血氧饱和度(pulse oxygen saturation,SpOb),使其维持在(90 ± 5)%。 开胸,朝着血流的反方向将弯形头皮针一端缓慢扎进肺动脉根部,头皮针另一端通过压力传感器连接生理通道记录仪。对肺动脉压力曲线进行记录。
1.3.3 肺损伤组织病理形态学观察
于缺氧12 d 给药后2 h,处死大鼠,取肺部组织,4%甲醛固定,脱水进行石蜡包埋,4 μm 进行组织切片,二甲苯脱腊,苏木精染,伊红乙醇染色,二甲苯处理,中性树脂进行封胶固定,烘箱过夜处理,光镜下观察肺组织切片病理学变化。
1.3.4 Western blot 法检测HIF-1α 和ET-1 在肺组织的表达
取“1.3.2”项下大鼠肺部组织,提取总蛋白,蛋白变性缓冲液变性,在硝酸纤维素膜上放置12.5%SDS-PAGE 胶转膜后,3% BSA 4℃封闭过夜,PBS 缓冲液洗涤硝酸纤维素膜,分别掺入HIF-1α 和ET-1单克隆抗体(1 ∶500),在室温下孵化45 min,PBS 缓冲液洗涤硝酸纤维素膜。 二抗孵育,掺入HRP 标记的二抗(1 ∶1000),室温孵化30 min,PBS 缓冲液洗涤硝酸纤维素膜,ECL 发光并成像,以β-actin 作为内参,检测HIF-1α 各组的相对表达,检测肺部组织中的ET-1 蛋白的相对表达量。 采用显微成像系统进行拍照,并对图片使用Image Pro Plus 6.0 进行处理分析,将染色阳性颗粒的灰度单位转换为吸光单位,并测量其密度(optical density,OD)值,以此作为其阳性表达的半定量。
1.3.5 ELISA 法检测血清中HIF-1α 与ET-1 的表达
各组大鼠于观察时间点采静脉血0.5~1.0 mL,3000 r/min 离心15 min,提取上清液,置离心管置于-20℃冰箱备用。 使用ELISA 试剂盒,严格按照说明书操作,计算血清HIF-1α 与ET-1 的含量。
1.3.6 定量PCR 检测肺组织的miR-155 mRNA表达
取出大鼠肺部组织研磨,加入1 mL TRIzol 裂解液,室温静置5 min,加入200 μL 氯仿混匀,室温放置10 min。 离心取上层,加入等体积异丙醇于RNase-free 的1.5 mL 离心管中,冰浴30 min,在4℃,12 000 r/min 离心15 min,得到RNA 沉淀,空气干燥RNA。 加入50 μL RNase-free 的水溶解RNA;利用NanoDrop 测定总抽提RNA,置于-20℃,备用。根据TaqMan® MicroRNA Reverse Transcription kit试剂盒操作说明书,cDNA 产物由抽提的RNA 体外反转录实验得到;在16℃中孵化需要30 min;在42℃中孵化需要30 min;85℃加热5 min,4℃保存。通过得到的cDNA 进行PCR 荧光定量检测,在LightCycler480 仪器上操作,以U6 为内参,miR-155的相对表达用ΔCT 来表示,95℃,预变性10 min,95℃15 s,60℃30 s,70℃30 s,共40 个扩增周期,ΔCT=CTU6-CTmiR-155计算miR-155 mRNA 的相对表达,引物信息见表1。
1.4 统计学分析
本研究中数据使用SPSS 22.0 进行分析,采用单因素方差分析计量资料以平均数±标准差(±s)表示,组间两两比较采用SNK-q 检验,以P<0.05 认为差异有显著性。
2 结果
2.1 各组大鼠的一般状况比较
模型组大鼠反应迟钝,虚弱,体毛暗淡,蜷缩少动,精神萎靡,食量减少和体重下降等;空白对照组大鼠反应敏捷,体毛光泽,饮食和体重正常;给药组大鼠的反应、体毛、饮食和体重介于空白对照组和模型组。
2.2 各组大鼠不同时间段mPAP 比较
与空白对照组比,模型组大鼠缺氧2、4、8 和12 d 的mPAP 显著增高(P<0.05),与模型组比,给药组大鼠缺氧2、4、8 和12 d 的mPAP 显著降低(P<0.05),见表2。
2.3 各组大鼠肺组织HE 染色
空白对照组肺组织结构清晰可见,间质无渗出,无炎症细胞浸润,肺泡腔内未发生水肿液与出血,肺泡壁完整;模型组肺微血管扩张充血,双肺体积增大,明显肺水肿,大量红细胞与液体从肺泡腔及肺泡隔内渗出,可见浸润的炎症细胞,偶然形成透明膜,大小各异的肺泡,肺泡间隔明显增厚;给药组大鼠其肺组织肺泡腔渗出、出血,毛细血管扩张开并充血,较模型组炎症细胞浸润明显减轻,见图1。
2.4 不同时间段各组大鼠血清中HIF-1α 与ET-1水平比较
与空白对照组比,模型组大鼠缺氧2 d、4 d、8 d 和12 d 的HIF-1α 和ET-1 均显著增高(P<0.05),与模型组比,给药组大鼠缺氧2、4、8 和12 d 的HIF-1α 和ET-1均显著降低(P<0.05),见表3 和表4。
2.5 各组大鼠肺组织ET-1 蛋白表达检测
与空白对照组比,模型组大鼠缺氧12 d 肺组织的HIF-1α 和ET-1 蛋白表达均显著增高(P<0.05),与模型组比,给药组大鼠缺氧12 d 肺组织的HIF-1α和ET-1 蛋白表达均显著降低(P<0.05),见图2 和表5。

表1 各引物序列信息Table 1 Primer sequence information
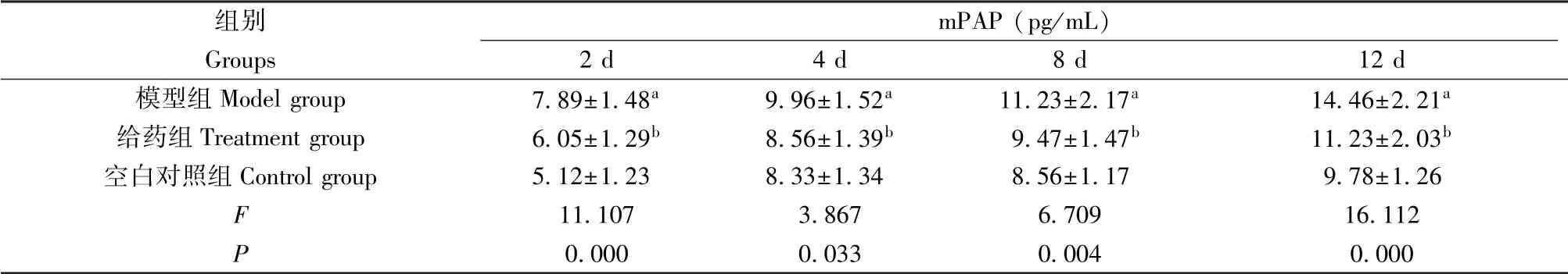
表2 各组大鼠不同时间段mPAP 比较(n=10)Table 2 Comparison of mPAP in the rats of different groups at different time of treatment (n=10)
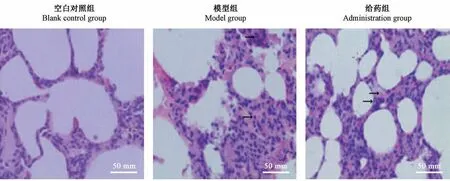
图1 各组大鼠肺组织的病理学改变Figure 1 Pathological changes in the lung tissues of rats in the three groups
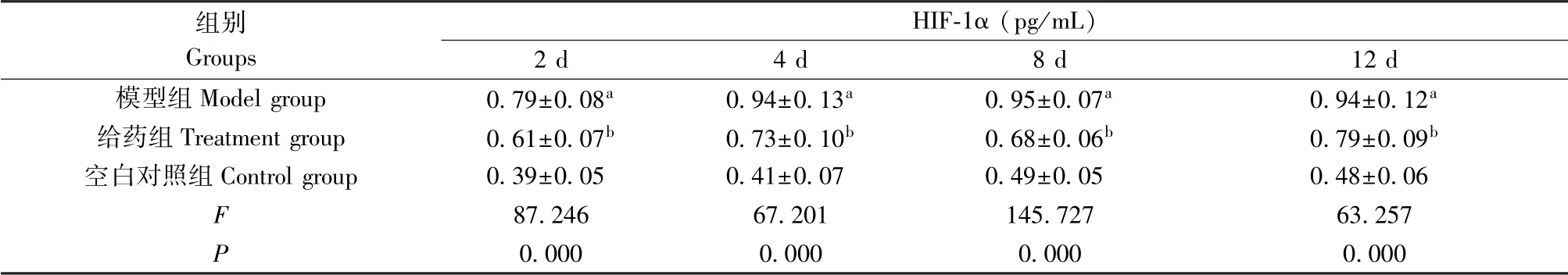
表3 不同时间段各组大鼠血清中HIF-1α 水平比较(n=10)Table 3 Comparison of the serum HIF-1α levels at different times of rats in the three groups (n=10)

表4 不同时间段各组大鼠血清中ET-1 水平比较(n=10)Table 4 Comparison of serum ET-1 levels at different times of rats in the three groups (n=10)
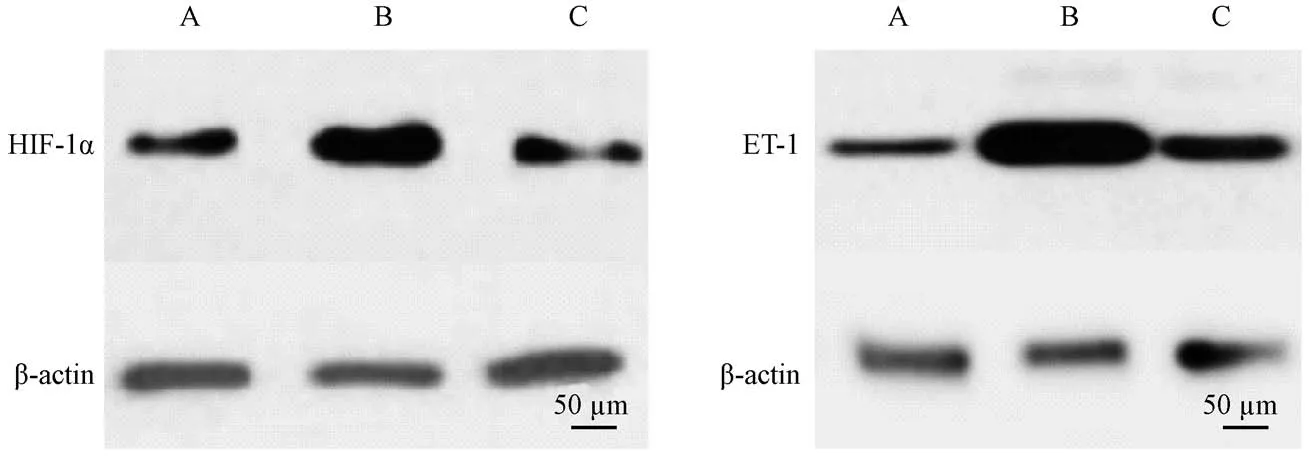
图2 Western blot 检测大鼠肺部组织中HIF-1α 与ET-1 的表达Figure 2 Western blot detection of the ET-1 expression in the rat lung tissues

表5 Western blot 检测大鼠肺部组织中HIF-1α 与ET-1 的表达(n=10)Table 5 Western blot detection of ET-1 expression in lung tissues of rats in the three groups (n=10)
2.6 各组大鼠肺组织miR-155 表达比较
与空白对照组比,模型组大鼠缺氧12 d 肺组织的miR-155 表达显著降低(P<0.05),与模型组比,给药组大鼠缺氧12 d 肺组织的miR-155 表达显著增高(P<0.05)。 进一步从TargetScan 网站对miR-155 的靶基因进行预测发现,其与HIF-1α 的5’UTR 配对互补,说明miR-155 可能是通过靶向结合HIF-1α 3’ UTR 调控其在肺组织中的表达水平。 见图3 和表6。

图3 各组大鼠肺组织miR-155 表达比较(n=10)Figure 3 Comparison of expression of microRNA-155 in lung tissue of rats in the three groups (n=10)
3 讨论
3.1 HPH 的发病机制及病理生理
HPH 的相关研究认为肺动脉高压的起始环节为缺氧所致肺微小动脉内皮损伤,细胞因子与血管活性物质产生及异常释放的原因为血管内皮受损导致功能失调引起[9]。 作用于血管平滑肌的血管舒缩因子平衡失调,出现肺血管收缩为早期表现,出现HPH,肺血管重建及肺血管壁病理改变为HPH后期表现[10]。 其中越来越受到关注的为HIF-1α 的作用[11],生理活性由α 亚基的表达和活性决定的氧依赖转录激活因子,是在缺氧条件下诱导产生的、在体内许多细胞中广泛存在的。 HIF-1α 是介导机体对缺氧发生基因表达重新调整、肺血管收缩、肺血管重塑形成的中心环节,在HPH 的发生过程中起着非常重要的作用[12]。 有研究表明缺氧早期低氧刺激肺组织产生大量HIF-1α,由于缺氧耐受效应使得肺组织HIF-1α[13]表达随着缺氧时间延长不再明显提高。 本研究表明,在HPH 大鼠模型中,早期缺氧刺激能显著提高HIF-1α 的表达,而在缺氧后8 d和12 d 虽然也有增加,但HIF-1α 的表达相比于前期无显著变化。
ET-1 是内源性血管收缩因子,肺部是ET-1 作用和代谢的最重要脏器[14]。 ET-1 可能是促使肺血管强烈收缩导致肺动脉高压发生过程中的一个重要因素[15]。 本次研究结果表明,在模型组新生大鼠缺氧2 d 起血清ET-1 即明显增高,并持续至12 d,同时在模型组大鼠肺组织中ET-1 蛋白也存在着显著高表达。 其原因可能为:在缺氧早期血管内皮细胞在低氧刺激下产生大量HIF-1α,HIF-1α 诱导下游靶基因ET-1 高表达。 肺血管内皮细胞膜上ET-B受体与通过自分泌途径异常升高的ET-1 相结合,生成大量氧自由基,对肺动脉高压损伤程度加重与肺血管内皮细胞本身的脂质造成过氧化损害[16]。?
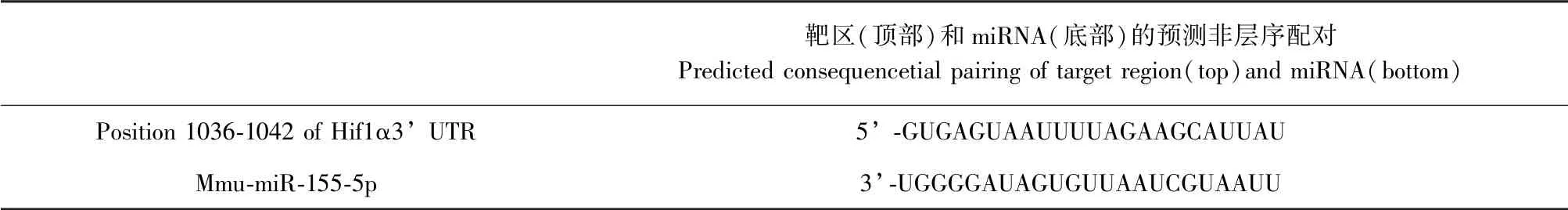
表6 miR-155 靶基因预测结果Table 6 Predictive results of target genes for microRNA-155
3.2 IGF-1 上调miR-155 表达对HPH 的作用
IGF-1 作为体内重要的生长因子,对细胞的增殖、分化、凋亡及机体的生长发育起重要调节作用[17-18];已有相关研究报道称IGF-1 可防治高氧肺损伤[19-20]。 在对HPH 大鼠模型进行现非侵入性鼻腔滴入IGF-1 后发现,肺部组织损伤明显降低,说明入IGF-1 对大鼠低氧肺动脉高压肺组织损伤有保护作用。 通过检测给药组大鼠肺组织HIF-1α 以及ET-1 的表达发现,其在肺组织中的表达水平得到了明显的抑制,表明IGF-1 可能通过作用于HIF-1α,进而抑制ET-1 的表达,发挥对肺组织的保护作用[21]。
IGF-1R mRNA 可在大鼠肺小动脉壁上正常表达。 IGF-1 可特异结合细胞膜ICG-1R,传导其刺激增殖信号,引发细胞增殖、细胞分化等效应。 ICG-1R 受激活后可影响大部分细胞的增殖和转化过程,同时抑制细胞凋亡[22]。 本次研究表明,模型组大鼠缺氧2、4、8 和12 d 的mPAP 平均水平为均显著高于空白对照组mPAP 对应时间的平均水平,各时间的差异均具有统计学意义,提示经低氧环境培养后,大鼠可产生低氧性肺血管重建和缺氧性肺动脉高压。 Dogansen 等的研究[23]表明,肺动脉高压的大鼠中,肺小动脉壁miR-155 水平明显减少。 分析原因,在低氧性肺血管重建过程中, IGF-1R 不仅担任受体角色,而且可引导肺血管平滑肌细胞从收缩表型转化成合成表型,促进细胞增殖。 而血管壁结构构成与细胞增殖及凋亡相关。 综上,IGF-1R 具有促进细胞增殖及抑制细胞凋亡的作用,因此有利于血管壁结构重建,而miR-155 是IGF-1R mRNA 中重要的抑制因子[24]。 因此,低氧条件时,肺小动脉壁miR-155 水平的降低与低氧性肺血管重建具有重要的关联,适当提升其水平有利于改善HPH 病情[25]。
在分子水平上采用定量PCR 检测肺组织中miR-155 的表达发现,与空白对照组大鼠比,模型组大鼠肺部组织中miR-155 表达明显下降,差异具有显著性。 与模型组大鼠比,IGF-1 给药后大鼠肺组织中miR-155 表达的平均水平则明显升高,差异有统计学意义。 对其靶基因进行预测分析发现,其可能通过靶向结合HIF-1α 5’UTR,调控其在肺组织中的表达。
低氧作为始动因素通过诱导HIF-1α 及其ET-1的表达,IGF-1R 具有促进细胞增殖及抑制细胞凋亡的作用,miR-155 对IGF-1R 的抑制作用减弱,导致肺血管收缩与增生异常,进而导致HPH 肺组织损伤。 IGF-1 可显著上调肺组织中miR-155 的表达,降低IGF-1R 的细胞增殖作用并促进细胞凋亡,一定程度上保护肺组织损伤的发生,对临床治疗缺氧性肺动脉高压引发的肺损伤提供理论依据。
猜你喜欢
实用癌症杂志(2022年12期)2022-12-26
山东第一医科大学(山东省医学科学院)学报(2022年8期)2022-12-07
新农业(2022年21期)2022-11-18
医学理论与实践(2022年21期)2022-11-10
中日友好医院学报(2022年4期)2022-10-15
天津医科大学学报(2021年3期)2021-07-21
中国食用菌(2020年9期)2020-11-11
看世界·学术下半月(2020年7期)2020-09-10
医学食疗与健康(2019年6期)2019-09-10
中华老年口腔医学杂志(2019年2期)2019-04-28
