
Spectrum of thalassemia mutations in fetuses of Han and Li ethinicities in Hainan province, China
2019-12-31 09:07ChaoLiangXueyinChenXueGaoHongjianChenYingxiaJinYaoZhouMinghongLiWencongWangWeiyingLuYuanhuaHuangJunWangQiLiYanlinMa
Chao Liang, Xue-yin Chen, Xue Gao, Hong-jian Chen, Ying-xia Jin,Yao Zhou, Ming-hong Li, Wen-cong Wang, Wei-ying Lu, Yuan-hua Huang, Jun Wang, Qi Li✉, Yan-lin Ma✉
1Hainan Provincial Key Laboratory for Human Reproductive Medicine and Genetic Research, the First Affiliated Hospital of Hainan Medical University, Hainan Medical University, Haikou, Hainan 570102, China
2Hainan Provincial Clinical Research Center for Thalassemia, the First Affiliated Hospital of Hainan Medical University, Haikou, Hainan 570102, China
3Prenatal Diagnosis Center, the First Affiliated Hospital of Hainan Medical University, Hainan Medical University, Haikou, Hainan 570102, China
4Key Laboratory of Tropical Translational Medicine of Ministry of Education, Hainan Medical University, Haikou, Hainan 571199, China
5Texas Heart Institute, 6770 Bertner Avenue, MC 2-255, Houston, TX 77030, USA
Keywords:Thalassemia Prenatal diagnosis Genetic diagnosis Amniotic fluid Genetic counseling
ABSTRACT Objective: To analyze the frequency and spectrum of thalassemia mutations in amniotic fluid samples collected from Han and Li people in Hainan province of China.Methods: We carried out a retrospective analysis on prenatal diagnosis of amniotic fluid samples collected from pregnant women who may have next generation with high risks of medium or severe thalassemia between 2005 and 2016. Diverse fetal thalassemia genotypes and mutated alleles in Han and Li people were analyzed and cmpared.Results: We examined 536 amniotic fluid samples from Han people and 588 from Li people,among which 406 Han and 500 Li samples were found to carry at least one thalassemia gene mutation, with a detection rate of 75.75% and 85.03%, respectively. Among all α- and β-thalassemia mutant alleles detected, the most frequently found mutations in Han and Li samples were SEA-type of α-thalassemia and 41/42 (-CTTT) of β-thalassemia, respectively.A total of 75 severe thalassemia cases were identified in Han samples and 53 in Li samples.In most of these severe cases, parents chose to terminate pregnancy after being informed of thalassemia-related risks.Conclusions: The thalassemia mutations shows ethnic and area specificity, and that prenatal diagnosis for high-risk thalassemia carrier pregnant women is an efficient approach to prevent and control the occurrence of severe thalassemia in the high-prevalence areas.
1. Introduction
Thalassemia, a group of autosomal recessive disorders characterized by the reduced synthesis of one or more normal globin chains, is one of the most prevalent inherited singlegene diseases[1,2]. A previous study has shown that thalassemia has highly specific molecular pathology and regional and racial/ethnic characteristics[3]. Geographically, thalassemia is fairly prevalent in the Mediterranean, Middle East, Indian subcontinent,Southeast Asia and South China[4,5]. In China, thalassemia is mainly prevalent in the southern cities and studies on the spectrum and genotypes of the mutations have been conducted in populations live in Yunnan, Guangxi, Guangdong provinces and Hong Kong of China[6-10]. According to the type of the globin molecule chain affected, thalassemia is divided into α-, β-, and δ-thalassemia, α- and β-thalassemia are more prevalent than δ-thalassemia. The majority of the mutations that cause α-thalassemia are deletions, whereas those causing β-thalassemia are generally point mutations[11]. In general, genetic diagnosis of thalassemia mainly aims to detect αand β-thalassemia.
Previous studies suggested that thalassemia had a high prevalence in Han and Li people[12,13]. As an ethnic minority group in China,most of the Li people live along the coast of southern China,especially in Hainan Island[14]. In Hainan province, the prevalence of thalassemia among Li is much higher than that of Han, the most frequently found α-thalassemia and β-thalassemia mutations are-α4.2, -α3.7, and 41/42 (-CTTT) respectively[15]. Different common mutated types have been found in different countries. For instance,previous reports showed that the most common mutation of α-thalassemia in Sardinia[16]was -α3.7and in the southern region of Vietnam was SEA-type mutation[17], and that the most common mutation of β-thalassemia in South Western Maharashtra was IVS I-5 (G-C)[18]and codon 26 (G>A) or Hb E (HBB: c.79 G>A) in South Vietnam[19]. It has been estimated that more than 330 000 infants affected by hemoglobin disorders were born annually, of which thalassemia accounts for 17%, suggesting that thalassemia presents an extremely serious global health burden[20,21].
Considering the high prevalence of thalassemia and its impact on patients’ life quality, prenatal diagnosis of the disease is enssential. A previous study on prenatal diagnosis of 1 058 pregnant women with β-thalassemia in Guangxi Zhuang Autonomous Region of China identified 315 normal fetuses, 500 carriers and 253 β-thalassemia major fetuses, among which pregnancies with severe β-thalassemia major fetuses were terminated after receiving patients’ written informed consent. Furthermore, the postnatal follow-up findings were consistent with the results from prenatal diagnosis performed on amniotic fluid[22]. Related data obtained from a study performed in Ahvaz, a city in Iran, showed that, among 316 samples evaluated by prenatal diagnosis, 180 cases (56.8%) were diagnosed as carrying at least one mutation in β-globin gene, suggesting that prenatal molecular diagnosis is effective in reducing the risk of hemoglobin disorders, especially in the regions of Iran with a high incidence of thalassemia[23]. Similarly, another study from Indore in India showed that, among 1 006 women screened by prenatal diagnosis during a 2-year period, 28 cases were diagnosed as having an abnormal pattern of β-thalassemia, and the prevalence of carriers was 2.78%[24]. In this study, as much as 99% of pregnant women with thalassemia mutations were willing to do prenatal diagnosis if requested[24]. Collectively, the aforementioned studies indicated that molecular test for prenatal diagnosis is an acceptable and efficient approach to identify the high-frequency mutations in thalassemiaprevalent regions and to reduce the number of newborns with severe thalassemia.
Prenatal diagnosis is undoubtedly the most effective way to prevent the birth of infants with severe thalassemia. As mentioned above,some studies on prenatal diagnosis and antenatal screening of thalassemia have been carried out in China[25,26]and other countries,such as India[27]and Iran[23,28]which mainly focused on its necessity in prevention of severe thalassemia birth and comparison of the invasive and non-invasive prenatal diagnosis or antenatal screening. Besides, studies on prenatal diagnosis reported the prevalence and genotypes of α-globin and β-globin gene mutations in Yunnan and Guangdong provinces of Southern China[29,30].Since thalassemia presented different variations of genes in different ethnic groups[31]. Previous studies only represented limited data on different genotypes of thalassemia of Han and Li samples in Hainan province. Since thalassemia is a common inherited disease in Hainan province, whose population is mainly composed of Han and Li people, performing prenatal diagnosis of Han and Li people is especially necessary. In this study, we analyzed the αand β-thalassemia genotypes from amniotic fluid samples collected from Han and Li people who visited our Prenatal Diagnosis Center from 2005 to 2016. To the best of our knowledge, this study was the first to analyze the spectrum of α- and β-thalassemia mutations of fetal samples from Li people based on prenatal diagnosis, and to compare it with those from Han people in Hainan province of China.This study will provide informative data for genetic counseling and prenatal diagnosis of thalassemia.
2. Materials and methods
2.1. Ethical approval
An ethical approval for undertaking this study was granted by the Ethics Committee of the First Affiliated Hospital of Hainan Medical University (No. 2017 research one). All participants signed the informed consent.
2.2. Participants
Between January 2005 and November 2016, A total of 1 232 fetal samples (amniotic fluid) collected from pregnant women that came for genetic counseling and prenatal diagnosis were detected for thalassemia in the Prenatal Diagnosis Center of Hainan Medical University. All these paticipants had been confirmed to carry a mutated α-globin and/or β-globin gene by polymerase chain reaction (PCR) test following abnormal routine blood test results that revealed hypochromic microcytic anemia or abnormal capillary electrophoresis. The detailed procedure of this study is shown in Figure 1. We defined the samples as Han if both parents were Han people, if at least one parent was Li people, the samples were considered as Li. A total of 108 samples from other ethnic groups, such as Miao, Zhuang and Hui, were excluded from this study. Finally this study included a total of 1 124 fetal samples including 536 samples from Han people and 588 samples from Li people. All the pregnant female carriers (age: 18-42 years old;gestational age: 17-28 weeks) took a prenatal genetic diagnosis of thalassemia using the molecular analysis of amniocytes.
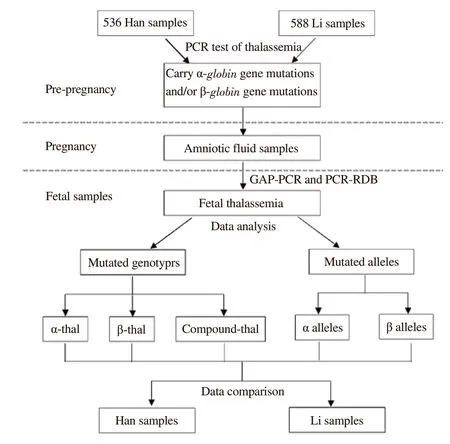
Figure 1. Flowchart shows the procedure of the study. All participants included in the study (536 Han and 588 Li) carried a mutated α-globin and/or β-globin gene diagnosed by PCR test in pre-pregnancy. These participants underwent prenatal diagnosis by amniocentesis during pregnancy because they may have next generations with high risks of medium or severe thalassemia. Amniotic fluid DNA was diagnosed by GAP-PCR and PCRRDB. Our study performed a statistical analysis and comparison according to diverse fetal thalassemia genotypes and mutated alleles in Han and Li participants.
2.3. DNA analysis of amniotic fluid
Fetal samplings ( amniotic fluid) were collected by amniocentesis under the guidance of ultrasound which is an invasive procedure.Genomic DNA was extracted from the amniotic fluid samples using thalassemia mutations test kit (Da’an Biotech, Guangzhou, China,and Yishengtang Biotech, Shenzhen, China). Gap-PCR was used to detect the deletion mutations of α-thalassemia (-α4.2, -α3.7, SEAtype). Polymerase chain reaction and reverse dot blot (PCR-RDB)was used to detect the point mutations of α-thalassemia, including αWS(Westmead), αQS(Quong Sze) and αCS(Constant Spring), and 17 common mutations of β-thalassemia, including codon 41/42(-CTTT), IVS-II-654 (C→T), -28 (A→G), codon 71/72 (+A),codon 17 (A→T), codon 26 (G→A), codon 31(-C), codon 27/28(+C), IVS-I-1 (G→T), codon 43 (G→T), -32 (C→A), -29 (A→G),-30 (T→C), codon 14/15 (+G), Cap (-AAAC), Int codon (ATG→AGG) and IVS-I-5 (G→C).
2.4. Data descriptions
The primary data of our study was the frequency and spectrum of thalassemia mutations in prenatal diagnosis fetal samples from Han and Li people in Hainan province. The detailed data collection and analysis process are shown in Figure 1.
2.5. Statistical analysis
A chi-square test was used to compare the data between different groups. P<0.05 was considered statistically significant. All data analyses were performed using SPSS 21 statistical software.
3. Results
3.1. Thalassemia mutations identified in Han samples
Among those 536 Han fetal samples tested, 406 (75.75%)had thalassemia mutations including 258 samples carried only α-thalassemia mutations, 106 samples carried only β-thalassemia mutations, and 42 carried both α-thalassemia and β-thalassemia mutations (Table 1).
Among those Han samples that carried only α-thalassemia mutations, 5 types of α-globin mutations including -α4.2, -α3.7, SEAtype, αWS, and αQS, were detected, in the form of 11 genotypes.Also, there were 170 samples with heterozygous mutations, and 88 samples with homozygous or compound heterozygous mutations(Table 1), which led to severe thalassemia. The most common genotype was Southeast Asia heterozygote (SEA-type/αα), followed by -α4.2/αα and SEA-type/SEA-type, with the rate of 17.91%, 6.72%and 6.53% respectively.
Among those Han samples that carried only β-thalassemia mutations, 6 types of β-globin mutations including 41/42 (-CTTT),-28 (A→G), IVS-II-654 (C→T), 17 (A→T), -29 (A→G), and 26 (G→A), were detected, in the form of 8 genotypes including 69 samples with heterozygous mutations and 37 samples withhomozygous mutations (Table 1). The three most common genotypes were 41/42 (-CTTT)/N, 41/42 (-CTTT)/ 41/42 (-CTTT)of and -28 (A→G)/N of, with the rate of 10.26%, 2.99% and 2.43%,respectively.
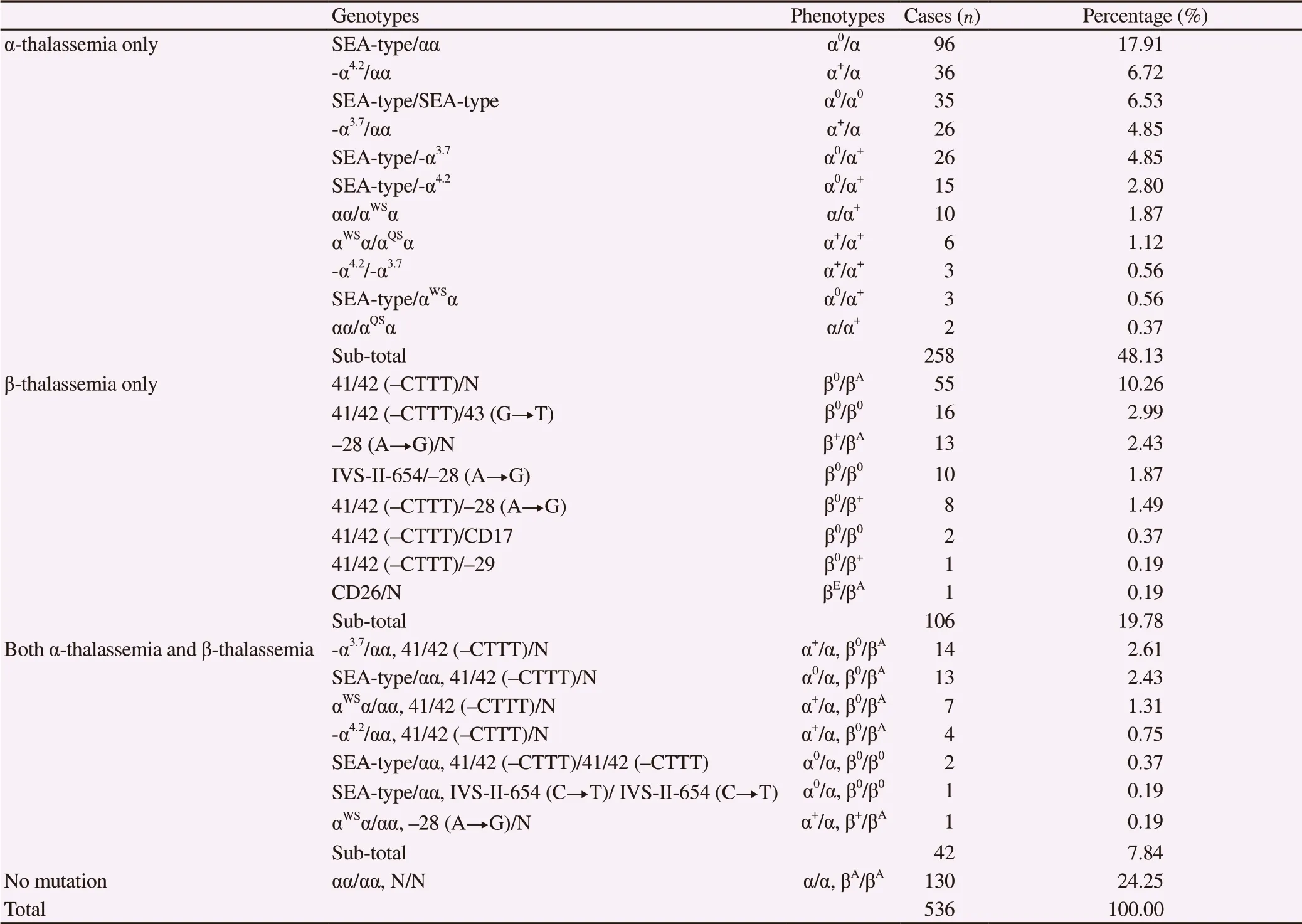
Table 1. Spectrum of α- and β-thalassemia genotypes detected in Han amniotic fluid samples.
Seven genotypes were found in Han samples that carried both α-thalassemia and β-thalassemia mutations (Table 1). The three most frequently found genotypes were -α3.7/αα, SEA-type/αα, and αWS/αα,all of which were accompanied by 41/42 (-CTTT)/N, with the rate of 2.61%, 2.43% and 1.31%, respectively.
Among all Han samples which carried thalassemia mutations, 75 cases had severe thalassemia, accounting for 13.99% of all tested Han samples. These cases included 35 cases of severe α-thalassemia(α0/α0) and 40 cases of severe β-thalassemia (β0/β0). In most of the severe cases, parents chose to terminate the pregnancy after they received the comprehensive information about thalassemia provided by the medical professionals.
3.2. Thalassemia mutations identified in Li samples
A total of 588 Li fetal samples were examined for thalassemia mutations, including 162 samples from couples where only one parent was Li (one-Li-parent), and 426 samples from couples where both parents were Li (two-Li-parents). Among these samples, 500 Li fetal samples (85.03%) were identified to carry thalassemia mutations, among which 126 were from one-Li-parent couples, and 374 from two-Li-parents couples, accounting for 77.78% (126/162)and 87.79% (374/426) of the examined samples respectively in each group (Table 2).
Similar to their Han counterparts, among Li samples carrying α-thalassemia mutations, 5 types of α-globin mutations (-α4.2, -α3.7,SEA-type, αWS, αQS) were found, but in the form of 14 genotypes.The most common genotype was -α3.7/αα (9.52%), followed by -α4.2/αα (7.48%) and αα/αWSα (4.93%). Five genotypes (-α4.2/αWSα, -α3.7/αWSα, αWSα/αWSα, -α3.7/αQSα, -α4.2/-α4.2) were only detected in two-Li-parents’ samples but not in one-Li-parent samples (Table 2),all of which were either homozygous or compound heterozygous mutations.
Only 2 types of β-globin mutations [41/42 (-CTTT), -28 (A→G)]were detected in Li samples. Among Li samples that carried only β-thalassemia mutations, three genotypes were detected (twoheterozygous and one homozygous), with 41/42 (-CTTT)/N being the most common one (7.48%).
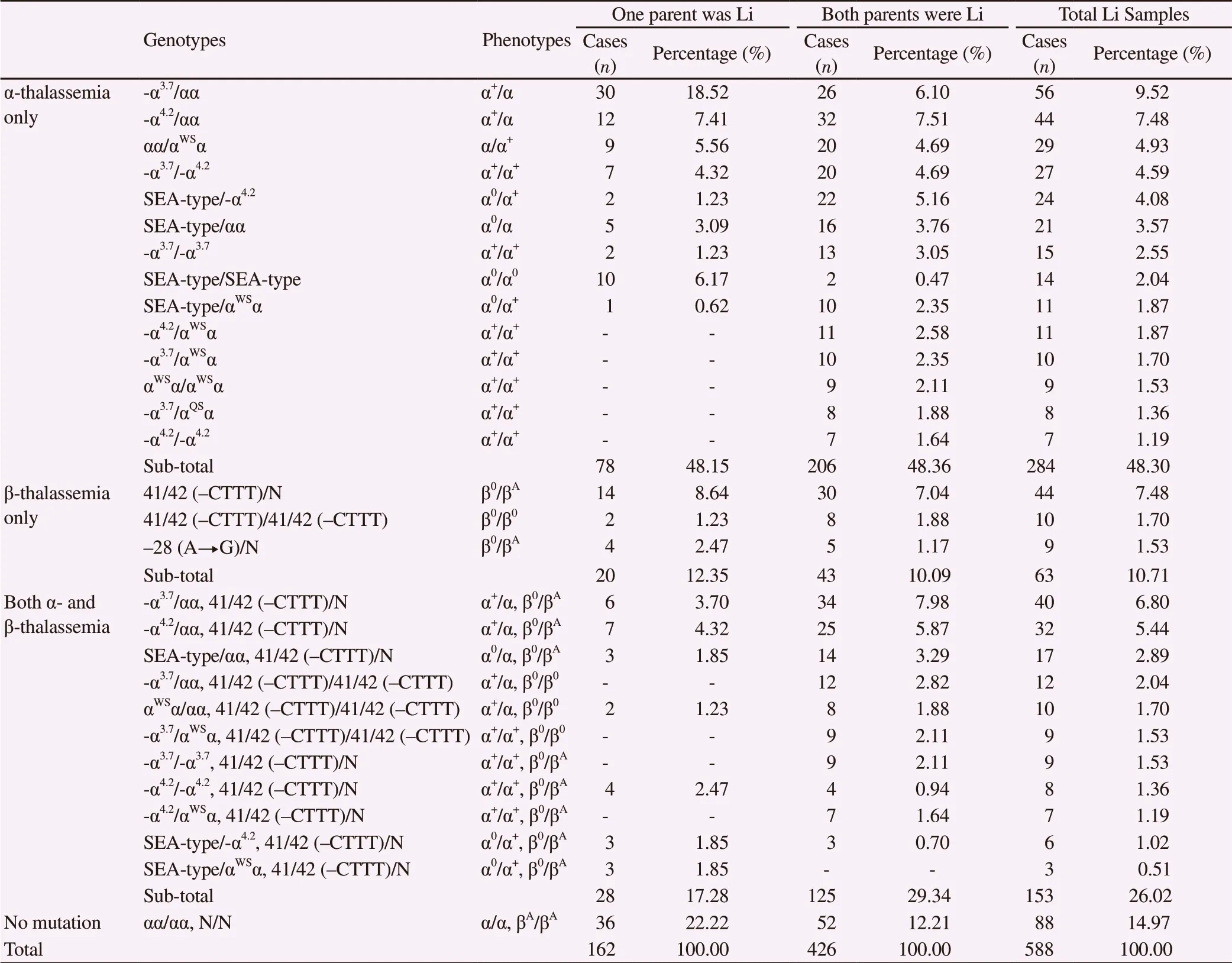
Table 2. Spectrum of α-thalassemia and β-thalassemia mutations detected in Li amniotic fluid samples.
In addition, among all Li samples that carried both α- and β-thalassemia mutations, eleven different genotypes were detected(Table 2), while seven types were found in one-Li-parent samples,and ten types were found in two-Li-parents’ samples. The three most frequently found genotypes were -α3.7/αα, -α4.2/αα, SEA-type/αα, all were accompanied by 41/42 (-CTTT)/N, with the rate of 6.80%,5.44% and 2.89%, respectively.
A total of 53 severe cases (9.01%) of thalassemia were identified in Li samples, including 12 cases of severe α-thalassemia (α0/α0) and 41 cases of severe β-thalassemia (β0/β0). Most severe cases in one-Liparent samples were α-thalassemia (10 cases of α-thalassemia and 4 cases of β-thalassemia); while most severe cases in two-Li-parents’samples were β-thalassemia (37 cases of β-thalassemia and 2 cases of α-thalassemia). Most parents of the severe cases chose to terminate the pregnancy after receiving the comprehensive information about thalassemia provided by the medical professionals.
3.3. Distribution of α- and β-thalassemia mutant alleles among Han and Li samples
Among all α- and β-thalassemia mutant alleles detected, the most frequently found mutation in Han and Li samples were SEA-type of α-thalassemia and 41/42 (-CTTT) of β-thalassemia, respectively(Table 3).
Five types of α mutant alleles were detected in the detected samples(Table 3). Among the Han samples, the most frequent α-thalassemia mutation was SEA-type, accounting for 58.25% of all α mutant alleles in Han samples. Among Li samples, the most frequent one was -α3.7, accounting for 34.04% of all α mutant alleles in Li samples. Comparison of the frequencies of the five α- mutant alleles between Han and Li samples showed significant difference.
On the other hand, six types of β mutant alleles were detected in Han samples, while only two types were detected in Li samples(Table 3). The most frequent mutation in both samples was 41/42(-CTTT), accounting for 72.09% in Han samples, and 96.50% in Li
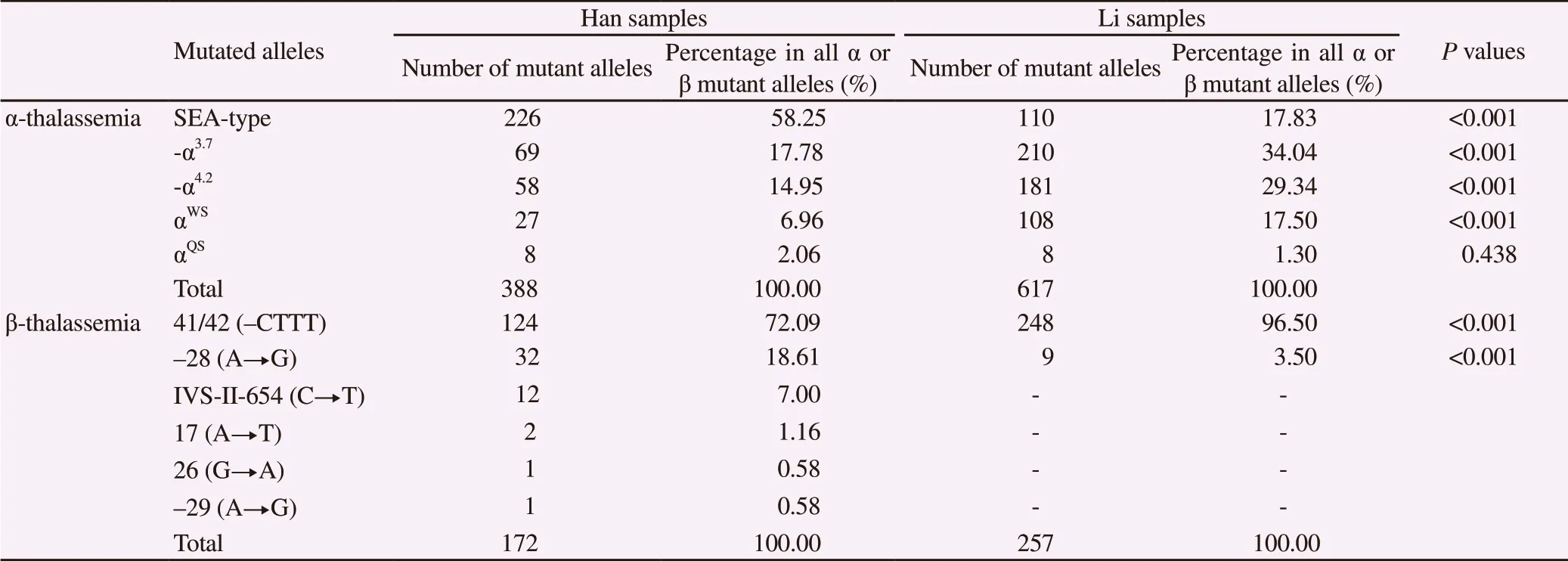
Table 3. Spectrum and frequency of α-thalassemia and β-thalassemia alleles in Han and Li amniotic fluid samples of Hainan province.
4. Discussion
In this study, we analyzed different fetal genotypes and frequencies of thalassemia mutations based on the prenatal diagnosis of Han and Li samples in Hainan province in China. Our study exhibited several advantages. Firstly, we have a relatively large sample size including 1124 fetal samples (amniotic fluid). Secondly, our data have valuable representativeness. Because our center has the largest resource for prenatal diagnosis in Hainan province, nearly all the carriers of thalassemia in the whole island come to our Prenatal Diagnosis Center. Furthermore, the peoples of Hainan province mainly consist of Han and Li, and the number of Li in Hainan accounts for the vast majority of all Li people in China. Therefore, our data represented a general feature of fetal thalassemia genotypes of Han and Li samples in Hainan province. Therefore, our study could reveal the different genotypes and frequencies of fetal thalassemia in Han and Li samples. In the present study, we described the nationality differencein fetal carriers of thalassemia mutations based on Han and Li samples in Hainan province.
In this study, we focused on not only the detectable rate but also the frequency and spectrum of α- and β-thalassemia genotypes in fetuses based on Han and Li samples. We observed that the detectable rates of fetal carriers with α- and/or β-thalassemia were different. Overall,the rates of thalassemia carriers were 75.75% and 85.03% among Han and Li samples, respectively. The frequency of α-thalassemia alone in Han samples was similar to that in Li samples (48.13%vs. 48.30%), whereas the frequency of β-thalassemia alone in Han samples was higher than that in Li samples(19.78% vs. 10.71%),and the frequency of compound thalassemia mutations in Han was obviously lower than that in Li samples (7.84% vs. 26.02%). In all Li samples, the major frequency difference in one-Li parent samples and two-Li-parent samples was also the compound thalassemia mutation (17.28% vs. 29.34%). Taking all together, our study indicated that α-thalassemia occurred most frequently and accounted for nearly 50% of all mutated genotypes in Han and Li samples.
Finally, the spectrum of α-thalassemia and β-thalassemia alleles showed obvious differences in Han and Li samples. SEA-type was the most frequent α-thalassemia mutant allele in Hainan Han samples. For Hainan Li samples, the most frequent α-thalassemia mutant allele was -α3.7(34.04%), followed by -α4.2(29.34%), SEAtype (17.83%) and αWS(17.50%). Regarding β mutant alleles, the mutant allele of 41/42 (-CTTT) was the most frequent one in these two samples. Even though the most frequent allele of β mutant alleles was 41/42 (-CTTT), the occurrence rate was different between Han and Li samples. And some mutated alleles were only detected in Han samples. Thus, we found that Li samples carried only 41/42 (-CTTT) and -28 (A→G), which was similar to the observations in Li adult samples from another study reporting only 41/42 (-CTTT) was detected[15].
In the present study, we found that Han samples still carried mutant alleles of IVS-II-654 (C→T), 17 (A→T), -29 (A→G) and 26 (G→A). Our statistical analysis indicated that 4 out of 5 α- mutant alleles between Han and Li samples were significantly different,and the distribution of 41/42 (-CTTT) and non-41/42 (-CTTT)alleles between Han and Li samples was also significantly different.In addition, we preliminarily compared the distribution of α- and β-thalassemia mutant alleles in Han and Li fetal samples with mutation distributions of fetal samples collected from Guangdong[32]and Guangxi[33]of Southern China where thalassemia also presented high incidences. We found the detection rate of -α3.7, -α4.2and αWSin Hainan Li samples was significantly higher than that in samples from Guangdong and Guangxi, and we also noticed that 71/72 (+A),IVS-I-1 (G→T), -43 (G→T) and Gγ+(Aγδβ)0were reported in research samples from Guangdong and/or Guangxi province but not in samples of Hainan province.
Based on our findings, the frequency of severe α-thalassemia in Han samples was significantly higher than that in Li samples, but the frequency of severe β-thalassemia was similar between Han and Li samples. The potential reasons underlying these differences were not clear, but it was possible that the unique national characteristics of Li, who has settled in Hainan Island for several thousand years,may contribute to these differences. For example, they mainly marry within Li people, which may increase the carrying rate of mutated alleles in the next generation. In addition, the carrying rate of thalassemia in Hainan province was similar with, or even higher in some mutant alleles than that reported in Guangdong and Guangxi provinces. Findings from several studies showed the protective effect of thalassemia against severe anemia in malaria patients in a number of countries, such as Thailand[34], Kenya[35], India[36], USA[37], and Maldives[38], it was likely that the high incidence of thalassemia may be partially attributed to malaria that once prevailed in Hainan Island.
According to the incomplete statistical data, it was estimated that currently more than 400 children suffer from thalassemia in Hainan province. Therefore, it is necessary to take measures of premarital screening, preconception test and prenatal diagnosis of pregnant women carrying thalassemia mutation(s) in Hainan province and other thalassemia-prevalent areas as well. A study from Cyprus confirmed the value of premarital screening policy, with a decline of more than 50% in terms of the thalassemia frequency among infants[39]. Those pregnant women who may have a fetus with severe thalassemia are often advised to terminate pregnancy by surgical or intravenous infusion of prostaglandin after prenatal diagnosis,which decreased the disability rate and improved the birth quality to some degree. However, the current prenatal diagnosis of thalassemia is not favored in some areas because of technical challenges, low income, ethical dilemmas and inadequate preventive programs[40-42].Fortunately, Hainan province has established the Prenatal Diagnosis Center, as well as a free screening program initiated by Health Department since 2011, Li Ka-shing Health Poverty Alleviation fund projects and Prevention and Control Committee established by Genetic Association of Hainan province, and Volunteers Association of Hainan province. All of these can ensure the prevention and thalassemia diagnosis as effectively and early as possible. In the past decade, the number of prenatal diagnosis for thalassemia in our Center has been increasing gradually from 8 cases in 2005 to over 300 cases in 2016.
Although this study only analyzed data of Han and Li samples, the results highlight the ethnic and regional specificity of thalassemia mutations considering Han and Li are the majority population live in Hainan, an island with high-incidence of thalassemia in China. We also believe that the prevention and control of thalassemia should come with an effective, accurate and accessible prenatal diagnosis.
Conflict of interest statement
We declare that we have no conflict of interest.
Authors’ contributions
C.L. developed data collection, data analysis and manuscript writing. H.J.C, Y.X.J, W.C.W. and Y.Z performed the samples preparation and PCR tests. Both X.Y.C and M.H.L provided cases collection and genetic counseling. And W.Y.L and Y.H.H also provided genetic counseling. Q.L and Y.L.M performed data analysis and manuscript review. X.G, J.W and Y.L.M were responsible for manuscript editing. And Y.L.M supervised the project.
 Asian Pacific Journal of Tropical Medicine2019年12期
Asian Pacific Journal of Tropical Medicine2019年12期
- Asian Pacific Journal of Tropical Medicine的其它文章
- Multi-targeting cytotoxic drug leads from mushrooms
- Seroprevalence of brucellosis among exposed agro-pastoral communities in southern Saudi Arabia
- Effect of El Niño Southern Oscillations on the incidence of enteric fever in Ahmedabad, India from 1985 to 2017
- Prevalence and risk factors of avian influenza H9N2 among backyard birds in Iran in 2015
- Alpinia officinarum Hance extract alleviates particulate matter-induced lung injury in mice
- Cutaneous leishmaniasis in an indigenous infant with Down's syndrome:A case report