
转录组测序在林草植物抗逆性研究中的应用
2019-12-20 02:41高慧娟吕昕培王润娟任伟程济南汪永平邵坤仲张金林
草业学报 2019年12期
高慧娟,吕昕培,王润娟,任伟,程济南,汪永平,邵坤仲,张金林
(兰州大学农业农村部草牧业创新重点实验室,兰州大学草地农业生态系统国家重点实验室,兰州大学草地农业科技学院,甘肃 兰州 730020,甘肃 兰州730020)
功能基因组学中的转录组学是从整体mRNA水平上研究特定组织或细胞在不同发育阶段,不同状态下功能基因的表达情况和调控机理,以阐明生物表型与功能[1-2]。自1977年Sanger法测序技术诞生,以末端终止法为核心的传统的第一代测序技术主导了DNA测序近30年,但是Sanger法测序通量低,准确度不高,价格昂贵,在应用过程中有很多缺陷[3-4]。随着科学技术的发展,DNA微阵列(DNA microarray)、DNA宏阵列(DNA macroarray)、基因表达系列分析(serial analysis of gene expression, SAGE)、大规模平行测序技术(massively parallel signature sequencing, MPSS)以及使用最广泛的第二代测序技术(next-generation sequencing technologies, NGS)即高通量测序技术(RNA-seq)相继产生[5-6]。二代测序技术自2005年被引入市场,逐渐替代了Sanger法并对基因组学的研究产生了深刻的影响[7]。相比于传统的测序方法,以Illumina/Solexa测序、Roche/454测序及ABI/SOLiD测序为主的RNA测序技术(RNA-seq)测序时间短、成本低、准确性高、高通量等,还可以获得低丰度的表达基因,亦可用于未知基因组序列的物种研究,因此被大规模的用于医学、生物学、农业等各个领域[8-11](表1)。该技术可以高通量的直接对mRNA反转录生成的cDNA进行测序,揭示特定细胞或组织中表达的全部转录本;通过不同处理间基因表达差异的比较,还可获得转录本表达量,确定转录发生位点,绘制转录图谱,预测一些基因模型和组蛋白修饰,进行遗传转化分析等[12-14]。基因组已知物种,转录组测序结果通过与其基因组比对,可以获得基因结构优化和基因可变剪切,发现新转录本,检测基因融合;无参考基因组的物种,通过测序组装(Denovoassembly)可获得单一基因序列,并通过功能比对获得基因信息,包括基因表达量及差异表达基因研究,还可检测转录水平的单核苷酸多态性(single nucleotide polymorphism,SNP)和简单重复序列遗传标记(simple sequence repeat,SSR)等[13-14]。近年来,RNA-seq在动植物发育调控, 免疫互作, 表观调控以及逆境响应上的应用也越来越广[15]。在植物抗逆方面,RNA-seq技术的应用能全面动态地检测逆境下植物体基因在各个时空下的表达变化,并挖掘功能基因,分析胁迫响应调控机制,为植物抗逆机理研究,培育抗逆品种提供分子遗传基础[16-17]。本研究详细总结了近年来国内外有关RNA-seq技术在林草植物抗逆方面的应用。

表1 DNA测序技术的发展
1 生物胁迫下林草植物转录组学研究进展
1.1 真菌性病害胁迫下林草植物转录组学研究
Barakat等[18]用454测序技术比较了栗疫病感染下美国板栗(Castaneamollissima)和抗病型中国板栗的基因表达情况。通过测序组装各自产生了28890和40039个Unigenes,GO注释表明大多基因都富集在生物与非生物刺激响应、细胞组织、生物进程、转移酶和水解酶代谢方面。栗疫病感染下一些与抗逆相关的转录因子和生物胁迫响应基因在中国板栗中发生了差异表达,其中有一些已被报道参与植物病原体防御反应,包括过敏性反应相关的细胞死亡以及阻止病原体进入细胞的物理屏障的构建,这些可能是导致两种板栗抗病性差异的主要原因。Wu等[19]利用Solexa技术对霜霉病感染下的葡萄(Vitisvinifera)叶片进行转录组表达分析,正常条件下获得了203514个Unigenes,霜霉病感染下获得了233653个Unigenes;通过基因表达分析发现,感病叶片中0.9%的Unigenes表达上调了5倍以上,0.6%的Unigenes表达下调5倍以上,且上调基因主要富集在核糖体结构、氨基酸和糖代谢通路中,这些差异基因以及代谢、信号通路可能与葡萄的抗病性密切相关。Wang等[20]利用Solexa测序技术研究了尖孢镰刀菌感染(0、2、4和6 d)下香蕉(Musanana)根部基因表达情况,组装获得了平均长度为1439 bp的25158个Unigenes,通过序列相似性比较,21622(85.94%)个Unigenes得到注释,真菌感染下与苯丙氨酸代谢、苯丙烷合成以及亚麻酸代谢途径相关的基因发生了差异表达,这些可能与香蕉抗病性密切相关。同年,Li等[21]利用RNA-seq技术比较了尖孢镰刀菌侵染下两种不同抗性香蕉(变异卡文迪香蕉和野生型香蕉)间的基因表达变化,共获得了平均长度为554 bp的88161个Unigenes,其中5008个基因与植物病害防御和信号转导相关;基因表达谱(gene expression profile,DGE)分析发现转录因子、细胞壁修饰基因以及与免疫激活、激素合成、病害防御相关的基因的表达在二者间有很大差异,导致了香蕉的抗病性的不同。稻瘟菌感染下水稻(Oryzasativa)转录组测序结果发现,参与信号转导和次生代谢反应的基因表达急剧上调,尤其是WRKY转录因子。已知OsWRKY的过表达可以增强水稻的抗病性,更加说明WRKY在水稻抗稻瘟菌反应中发挥重要作用[22]。轮状镰刀菌会引起玉米(Zeamays)穗腐病,降低籽粒产量。Wang等[23]对镰刀菌有较高抗性的玉米柱系进行了RNA测序分析,发现热激蛋白类、次级代谢物(苯丙酸、木质素、香豆酸、类黄酮)以及脱落酸、茉莉酸、水杨酸信号通路在玉米抵抗真菌感染过程中发挥重要作用。Sun等[24]对水稻条锈病感染下的拟南芥(Arabidopsisthaliana)进行了RNA测序分析,总共检测到624个差异表达基因,在感染14 d后,有255个基因上调,38个下调,感染21 d后,146个基因上调,237个下调,其中只有52个基因在整个感染期间均差异表达。即在感染早期(14 d),与宿主防御反应相关的基因被诱导表达,而到后期(21 d)表达被抑制。功能注释发现这些基因与植物防御反应、次生代谢及蛋白质磷酸化相关。研究发现大白菜(Brassicapekinensis)R635-10不易感染十字花科根肿病菌,而S177-47极易感染根肿病菌,Jia等[25]对这两个品种进行了RNA-seq分析,发现R635-10感染病菌后,有2089个基因差异表达,其中与钙离子内流、葡糖异硫氰酸盐合成、细胞壁增厚、水杨酸稳态及几丁质代谢相关的基因都上调表达。S177-47感染病菌后有10038个基因差异表达,其中与三羧酸循环、DNA复制、氧化磷酸化、细胞壁扩张、根瘤素、吲哚乙酸和细胞分裂素相关的基因上调表达,这些基因主要参与了细胞周期控制、细胞分裂及能量合成和转换过程;而细胞分裂和扩张基因在R635-10中被抑制表达,说明在S177-47中根细胞分裂失去调控导致了根的肿胀。
1.2 细菌性病害胁迫下林草植物转录组学研究
Martinelli等[26]通过RNA-seq研究黄龙病感染下柑橘(Citrusreticulata)的转录组变化情况。黄龙病感染下,几个WRKY转录因子、与光合作用光反应、ATP合成、乙烯通路、蛋白降解和错误折叠相关的基因表达上调;而与细胞分裂素和赤霉素合成以及信号转导相关的基因表达受到抑制。通路分析发现,与源库运输相关的蔗糖和淀粉代谢,以及激素合成和信号通路发生了改变。这些结果说明了黄龙病感染下柑橘的基因响应情况,为以后减缓发病提供了广泛的数据基础。龙葵(Solanumnigrum)对青枯雷尔氏菌引起的青枯病有较强的抗性,Zuluaga等[27]对青枯病感染下的龙葵F118(抗病)和F97(易感病)进行了RNA测序分析,在F118和 F97中分别有221和644个基因发生差异表达,其中上调表达基因中有1/2已被报道参与植物病原反应。乙烯和水杨酸相关基因只在F118中被诱导表达,茉莉酸相关基因在F118和 F97中均表达下调,这为以后龙葵抗青枯病研究提供了重要的分子基础。根际有益菌能提高植物对病原菌的抵抗力,Van de Mortel等[28]对荧光假单孢菌SS101 (Pf. SS101)在拟南芥抵抗细菌性斑点病反应中的功能进行了分析。发现荧光假单孢菌能够增强植株对细菌性斑点病的抗性,通过转录组测序发现,其他根际细菌一般通过乙烯信号来提高植物防御能力,而Pf. SS101是依靠水杨酸信号来提高植物抗病能力,尤其葡糖异硫氰酸盐和植保素是Pf. SS101诱导植物抗细菌性斑点病必不可缺的次级代谢物。
1.3 虫害胁迫下林草植物转录组学研究
大豆包囊线虫(soybean cyst nematode, SCN)寄生于豆类植物根部,对豆类生长和种子产量造成严重危害。变异山羊草(Triticumtriunciale)对包囊线虫具有一定的抗性,由于基因组信息的缺乏,相关抗虫功能基因研究受到限制。Xu等[29]运用RNA-seq对包囊线虫感染下的山羊草根部组织进行测序,获得了平均长度为500 bp的118064个Unigenes。功能分类发现7408个Unigenes与植物防御和抗性有关,包括“植物病原互作”“磷脂酰肌醇信号系统”和“ABC 转运体”,这为抗虫基因筛选以及山羊草抗包囊线虫分子机制研究提供了一定的基础。Jain等[30]对SCN感染下的两种黑白斑豆(Glycinemax), PI533561(抗SCN)和GTS-900(易感染SCN)进行了RNA测序分析,发现PI533561感染SCN后有353个基因发生差异表达,其中154个上调表达,GTS-900感染SCN后有990个基因发生差异表达,406个上调表达。其中,WRKY、病程相关蛋白和热激蛋白基因均发生差异表达,光合反应均被抑制。黑豆(Glycinemax)对SCN具有较强的抗性,Li等[31]通过RNA测序分析了灰皮支黑豆对SCN的抗病机制。发现在SCN侵染5、10和15 d后灰皮支黑豆根部分别有740、1413和4925个基因发生了差异表达,其中有225(3个下调)个基因在整个时间段均差异表达。且大多数基因与植物防御以及激素信号相关,为了验证激素信号在灰皮支黑豆抵抗SCN中的作用,对灰皮支黑豆进行了生长素、赤霉素、水杨酸、茉莉酸以及乙烯处理,结果发现激素处理能够显著提高灰皮支黑豆对SCN的抗性。芽孢杆菌Sneb545能提高大豆(Glycinemax)对SCN的抗性,Kang等[32]通过转录组测序对经过Sneb545处理的大豆响应SCN的防御机制进行了分析,发现与未处理的大豆相比,Sneb545处理的大豆中有大量的杀线虫代谢物,包括4-乙烯基苯酚、蛋氨酸、胡椒碱和棕榈酸,这些代谢物增强了大豆的抗病性。
1.4 病毒性病害胁迫下林草植物转录组学研究
Rubio等[33]利用Solexa技术分析李痘病毒感染下桃树(Amygdaluspersica)叶片的基因表达变化。没有症状发生的叶片,初期伴随有腺苷甲硫氨酸的产生,且与茉莉酸、几丁质酶以及细胞分裂素等相关的基因发生差异表达。有痘病症状的样品中,有一部分基因比对到病毒参考基因组。GO注释表明与生物刺激响应,脂类和碳水化合物代谢有关的基因发生了差异表达。此次结果阐明了转录水平下桃树感病和抗病的分子机制的不同。马铃薯(Solanumtuberosum)病毒病对马铃薯产量造成严重影响,不同马铃薯品种对病毒病的响应有很大差异。Goyer等[34]通过RNA测序比较了两个马铃薯品种Premier Russet和Russet Burbank在病毒病(PVY)感染下的基因表达变化。Premier Russet栽培种对PVYO有抗性,但易感染PVYNTN,Russet Burbank易感染PVYNTN和PVYO。结果发现感染4 d后,大多数基因下调表达,感染10 d后,大多数基因上调表达。Premier Russet感染PVYO反应中,差异表达基因大多与光合作用中的光捕获相关,Premier Russet感染PVYNTN和Russet Burbank感染PVYO反应中,差异表达基因大多与核小体组装有关。这些基因中,ABC转运蛋白、MYC2 、脂质转运蛋白及木葡聚糖内转糖苷酶/水解酶基因只有在Premier Russet感染PVYO反应中下调表达,说明这些基因的表达决定了马铃薯对PVY的抗病能力的强弱。Rodamilans等[35]对李痘病毒感染下的欧洲李(Prunusdomestica)进行了RNA测序分析,总共有3020个基因发生差异表达,其中与植物防御相关的基因大多都上调,而光合相关基因都下调表达。上调基因中有10个是核苷酸结合位点-富含亮氨酸重复基因(NBS-LRR),NBS-LRR在文献中多有报道参与植物抗病反应[18,21,26]。
总的来说,通过RNA测序,我们可以分析病虫侵害下植物基因表达变化,从而筛选出一些病虫害响应基因;也可以通过比较两个不同抗性品种在生物胁迫前后的基因表达差异,直接鉴定出病虫害防御基因,探究抗病机理。植物对真菌、细菌、虫害以及病毒的响应机理大多是相通的,尤其一些苯丙氨酸、蛋氨酸、甲硫氨酸、转录因子(WRKY)、激素(脱落酸、茉莉酸、水杨酸、乙烯)、热激蛋白、次级代谢物(苯丙酸、香豆酸、类黄酮、胡椒碱、棕榈酸、葡糖异硫氰酸盐和植保素)、NBS-LRR、细胞壁修饰基因(几丁质和木质素)以及植物防御相关基因在植物应对生物胁迫过程中发挥重要功能。
2 非生物胁迫下林草植物转录组学研究
2.1 盐胁迫下林草植物转录组学研究
胡杨(Populuseuphratica)是一种耐盐模式植物,Qiu等[36]利用Solexa技术对盐处理前后(100 mmol·L-1NaCl)胡杨进行转录组测序分析,组装获得了86777个Unigenes,27%的Unigenes发生差异表达,且它们主要参与物质转运、转录调控、细胞信号转导、脱落酸(abscisic acid,ABA)调节以及生物合成等反应,这为以后胡杨盐胁迫响应的分子机制研究奠定了基础。Chen等[37]利用Solexa技术分析高耐盐红树植物杯萼海桑(Sonneratiaalba)在盐处理下(250 mmol·L-1NaCl)的转录组表达情况, 组装获得了平均长度为581 bp的30628个Unigenes,鉴定了2358个SSR标记,基因功能注释发现与线粒体、碳代谢和次生代谢相关的基因参与了海桑的盐适应过程。另外,通过与已知的273个其他物种中的盐响应基因比对,在海桑中筛选出了1266个的耐盐候选基因。将杯萼海桑转录组序列与其他同种海桑的表达系列标签(expressed sequence tags, EST)比较,鉴定了4个具有较强自然选择标记的基因,这为海桑耐盐机理及遗传多样性研究提供了丰富的理论依据。Chen等[38]利用Solexa和DGE技术分析盐处理下小叶杨(Populussimonii)的转录组表达变化,在3、6和9 d的盐处理下分别检测到了5453、2372和1770个差异表达基因,GO功能分析表明大多数差异表达基因与物质运输和逆境响应相关。Huang等[39]利用Solexa测序技术研究盐处理下半红树植物水黄皮(Pongamiapinnata)的转录组基因表达情况,获得了平均长度为606 bp的108598个Unigenes,54596(50.3%)个Unigenes获得注释,盐处理下检测到了23815个显著差异表达基因,这些基因的功能富集分析显示出组织特异性,根中的差异表达基因数多于叶中。2013年,Ma等[40]又利用RNA-seq技术进行了盐处理下(200 mmol·L-1NaCl)胡杨根、叶和毛白杨(Populustomentosa)愈伤组织的基因表达分析,分别检测到 6727、3954和3733个差异表达基因,许多与阳离子运转以及氧化还原酶相关的基因在胡杨中特异表达,这可能导致了二者之间的耐盐性差异。两个甜瓜(Cucumismelo)品种玉露和冰雪脆在300 mmol·L-1NaCl下的生理参数存在差异,陈嘉贝等[41]利用Solexa测序技术检测它们在NaCl处理下的基因表达情况,发现玉露共有56个转录因子基因发生差异表达(22个上调表达,34个下调表达),冰雪脆有47个转录因子发生差异表达(17个上调表达,30个下调表达)。转录因子表现出一定的品种特异性,两品种间也有部分重叠。赵航等[42]利用RNA-seq技术分析盐处理下(600 mmol·L-1NaCl)盐穗木(Halostachyscaspica)基因表达情况,并分析其相应的响应方式。研究获得了大量的差异表达基因,通过分类汇总,得到23组与耐盐相关的基因类群,包括柠檬酸合成酶、蛋白激酶、WRKY成员等,这与盐穗木的耐盐性密切相关。Joann等[43]利用RNA-seq技术研究盐胁迫下碱蓬(Suaedaglauca)的转录组表达变化,组装获得了平均长度为763 bp的54526个Unigenes。基因差异表达分析检测到475个Unigenes表达下调,44个上调。功能分析发现许多基因与离子转运、信号调节、细胞代谢以及干旱和耐盐响应相关,此次结果鉴定了许多参与碱蓬耐盐反应的候选基因,也为其他多肉植物研究提供了丰富的参考基因序列。高彩婷等[44]利用Solexa和DGE技术检测300 mmol·L-1NaCl处理下VAO-9燕麦(Avenasativa)叶片在0.5、3、8和24 h下的基因表达情况,并测定了相对电导率、丙二醛和脯氨酸含量变化。结果获得了平均长度为645 bp的65810个Unigenes。基因差异表达分析检测出0.5 h有306个基因表达上调,64个下调;3 h下639个上调,290个下调;8 h下1488个上调,882个下调。通路分析发现差异基因大多富集在激素信号转导途径、磷酸肌醇途径以及渗透调节等途径。而且胁迫下生理指标的变化与相对应的Unigenes的表达变化一致。Ma等[45]利用RNA-Seq技术分析了旱生植物霸王(Zygophyllumxanthoxylon)在盐和渗透胁迫下的转录组表达情况,组装获得了平均长度为680 bp的106423个Unigenes。胁迫下检测到与Na+、K+、Ca2+、Mg2+、氮、磷酸盐以及微量元素转运,离子区划及活性氧清除相关的基因显著上调。且相较于渗透胁迫,盐胁迫下与活性氧清除、光合电子传递及碳固定相关的基因明显上调,而叶绿素分解相关基因表达减少。这些结果为以后牧草类以及农作物抗逆机制研究提供了丰富的基因资源。An等[46]利用RNA-seq技术分析盐碱胁迫下紫花苜蓿(Medicagosativa)的转录组表达变化,共获得了53853个Unigenes,处理第1天2286个基因发生差异表达,第7天2233个基因差异表达。大多数基因与过氧化物酶、光反应、类黄酮代谢、脂质代谢以及转录因子相关。另外,转录因子NAC和谷胱甘肽转移酶基因在盐胁迫表达下调,而在盐碱胁迫下表达上调,这为苜蓿耐盐碱机制研究提供了新的思路。250 mmol·L-1NaCl处理下,对红麻(Hibiscuscannabinus)进行RNA测序获得了2384个差异表达基因(1702个上调),GO富集发现抗氧化酶类基因均上调表达,大多数基因与氨基酸代谢以及碳水化合物代谢通路相关[47](表2)。综上所述,通过转录组测序能够发现转录因子WRKY、抗氧化酶类、蛋白激酶以及与离子转运、类黄酮代谢、ABA信号以及碳固定相关基因在植物应对盐胁迫过程中发挥重要功能。
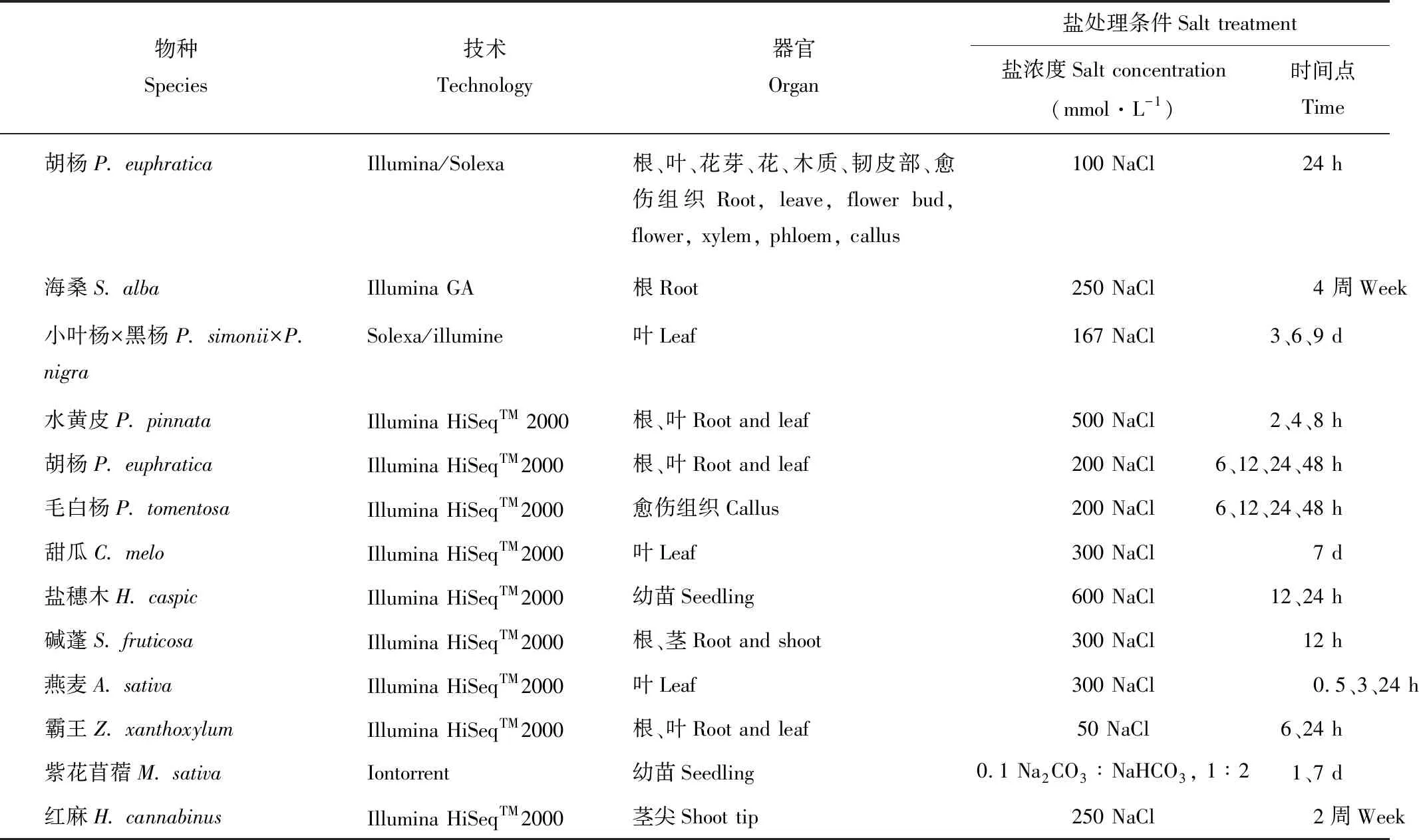
表2 部分植物耐盐分子机制的转录组学研究
2.2 干旱胁迫下林草植物转录组学研究
Zhou等[48]利用454测序技术分析沙冬青(Ammopiptanthusmongolicus)根在PEG处理下的基因表达差异,组装获得了15173个contigs 和13883个singlets,检测到1827个SSR分子标记,功能分析发现了数百个干旱胁迫相关基因,为以后沙冬青耐旱机制研究奠定基础。Yu等[49]利用Solexa测序技术研究干旱胁迫下(0、1、2和3 d)大白菜(Brassicapekinensis)基因表达情况,通过测序比对分别有13036、12472、12774和12227个特异性标签比对到数据库,基因表达分析发现胁迫下总共有1092个基因发生显著差异表达,包括37个转录因子基因、61个渗透响应基因和28个信号转导相关基因。Chen等[50]利用RNA-seq技术研究干旱处理下2个棉花(Gossypiumhirsutum)品种(耐旱型和干旱敏感型)的基因表达情况,发现有13.38%~18.75%的Unigenes发生差异表达,且随着处理时间的延长,差异表达基因数目增多。基因表达分析发现干旱处理下耐旱型棉花的一些包括光合作用在内的细胞代谢过程被抑制。两种棉花品种中与ABA信号、乙烯信号以及茉莉酸信号通路相关的基因表达水平存在显著差异,说明这些信号通路可能参与棉花抗旱反应。Shi等[51]利用Solexa技术分析干旱胁迫下C3旱生灌木红砂(Reaumuriasongarica)的转录组表达变化并鉴定其相应的耐旱基因,组装获得了平均长度为677 bp的77647个Unigenes,其中43054个能比对到已知蛋白,差异基因分析检测到123个抗旱相关的候选基因,C4植物光合作用相关基因在红砂中也有所表达,这为旱生植物基因功能分类以及荒漠植物适应干旱环境的遗传基础研究提供了丰富的基因资源。Long等[52]利用Solexa技术分析干旱处理下梭梭(Haloxylonammodendron)转录组基因表达情况,组装获得了平均长度为728 bp的79918个Unigenes,25619个在Nr数据库获得注释。基因表达分析发现干旱处理下356个基因上调表达,704个下调表达。通路分析发现部分差异表达基因与脂肪酸、淀粉和氮代谢相关,这些代谢途径可能参与梭梭干旱响应过程;并检测到了35个干旱诱导的转录因子,包括WRKY、MYB和bZIP家族成员,这为逆境下荒漠植物基因和基因组研究提供了丰富的基因基础。Steven等[53]利用RNA-seq技术研究干旱下两种红三叶草(Trifoliumpratense)的干旱响应机制,组装获得了45181个contigs,鉴定了3127个SSR位点,在7178个contigs中检测到27922个稀有等位基因SNP。而且正常条件下许多衰老相关基因都在敏感型红三叶草中高丰度表达,干旱处理下干旱敏感型红三叶草中差异表达基因数目大约是耐旱型红三叶草的2倍,这为红三叶草的耐旱机制研究奠定了丰富的基因基础。Xiao等[54]分析了不同脱水情况下牛耳草(Boeahygrometrica)的基因表达情况,发现1239 个基因特异性响应中度脱水,4135个基因特异性的响应重度脱水;且与植物-病原互作和激素相关的基因在不同脱水情况下均差异表达,而与mRNA检测通路,尤其是光合作用和氮代谢相关基因只在重度脱水下差异表达,其中ABA代谢和信号、磷脂信号、胚胎发育晚期蛋白、过氧化物酶以及早期光诱导蛋白基因大多数都上调表达,这为牛耳草耐旱机制研究提供了丰富的理论依据。Zhu等[55]对PEG处理下的一年生禾本科牧草苏丹草(Sorghumsudanense)进行了基因表达分析,在短期的PEG处理下(3 d),获得了2329个差异表达基因,大多与碳固定和激素信号通路相关。在长期的PEG处理下(6 d),获得了5101个差异表达基因,大多与激素信号通路相关,还检测到17548个SSR。本课题组利用RNA-seq对-0.75 MPa 渗透胁迫处理下的梭梭干旱响应机制进行了分析,在梭梭地上部和地下部分别产生了平均长度为556 和535 bp的82736 和99624个 Unigenes;种类分布发现,梭梭与葡萄同源性最高,同时鉴定到了13486个 SSRs;GO功能分类发现大多数基因与细胞代谢及催化活性相关;COG分类发现多数基因与转录、翻译以及核糖体结构相关;KEGG分类发现多数基因与翻译及碳水化合物相关。DGE分析发现,与对照相比,渗透胁迫下地上部分总共有3353个差异表达基因,其中627个基因在6和24 h下均差异表达;地下部分总共有4564个差异表达基因,其中821基因在6和24 h下均差异表达。地上部与光合、寡糖、有机酸、胺、异戊二烯以及质子转运相关的基因下调表达,而与胁迫、水分匮缺以及茉莉酸刺激响应、水杨酸代谢以及类黄酮和乙烯合成相关基因上调表达;地下部分与分生组织生长、色素积累调节以及根系发育相关基因下调表达,与水杨酸信号通路、植物螯合肽代谢、激素刺激、氧化和高渗透胁迫响应相关基因上调表达。本研究又进行了干旱响应基因筛选,发现与离子转运、活性氧清除、细胞壁和细胞膜结构维持、木质素、有机渗透调节物以及激素合成相关的大多数基因均上调表达,还有一些常见的水通道蛋白、胚胎发育晚期蛋白(包括脱水素和脱落酸胁迫成熟诱导蛋白)以及热激蛋白基因都上调表达,这为梭梭耐旱机制研究提供了丰富的分子基础,也为植物抗旱分子机制研究以及优良牧草和作物培育提供了大量的候选基因[56](表3)。综上所述,通过转录组测序发现转录因子(WRKY、MYB和bZIP)、水通道蛋白、胚胎发育晚期蛋白、热激蛋白、激素信号(ABA、乙烯以及茉莉酸信号)以及活性氧清除相关基因在植物应对干旱胁迫过程中发挥重要功能。
2.3 低温及高温胁迫下林草植物转录组学研究
Zhao等[57]利用Solexa测序技术对生长于阿尔卑斯山具有极强耐冷性的高山离子芥(Chorisporabungeana)进行了低温处理下的转录组表达分析,并与拟南芥进行比较。结果获得了平均长度为888 bp的54870个Unigenes,基因表达分析检测到3484个基因表达上调,4571个表达下调。还发现了一个与离子芥和拟南芥耐寒相关的生长调节因子Karrikins。功能富集分析发现与低温适应相关的基因在拟南芥中上调表达,包括转录因子CBF2和CBF3,而在高山离子芥中没有CBF同源基因的差异表达,说明低温适应不是离子芥主要的耐冷方式;但是与蛋白磷酸化和自动泛素化作用相关的基因在高山离子芥中高丰度表达,使它能够快速,灵活的适应寒冷环境,这也导致了拟南芥和离子芥耐寒机制的不同,这些结果为植物耐低温关键基因挖掘提供了重要的理论依据。Tian等[58]利用Solexa测序技术研究低温诱导下火鹤花(Anthuriumscherzerianum)转录组差异基因表达情况,组装获得了平均长度为560 bp的44382个Unigenes,其中27396个能比对到已知蛋白。总共检测到了4363个差异表达基因,1、5和 24 h下分别有292、805和708个基因上调表达。KEGG通路分析发现1 h下差异表达基因主要富集在光合、代谢以及氧化磷酸化途径;5 h下主要富集在氧化磷酸化途径;24 h下主要富集在mRNA检测、RNA转运以及植物病原互作途径。还发现了39个低温诱导相关的转录因子包括AP2/ERF、Zinc finger protein、NAC、MYB和bZIP 家族成员,这些为火鹤花基因和基因组研究提供了丰富的基因资源。Xin等[59]利用RNA-seq技术研究耐寒型长白山葡萄(Vitisvinifera)的抗寒响应机制,并与欧洲葡萄进行比较。发现在低温处理下,长白山葡萄差异表达基因数(1314)少于欧洲葡萄(2307),有408个基因在二者中均差异表达,在葡萄抗寒反应中起基础防御作用。功能分类表明长白山葡萄含有较多的与代谢、信号转导和转录相关的上调基因,这些基因可能在葡萄抗寒反应中发挥重要作用。Wang等[60]利用Solexa测序和DGE技术研究低温下茶树(Camelliasinensis)的基因表达变化,并分析其响应方式。结果获得了平均长度为356 bp的216831个Unigenes,1770个基因发生了差异表达(1168个上调),大多与信号转导、低温响应、质膜稳定性以及渗透响应等相关。通路分析表明碳水化合物代谢和钙信号通路可能在茶树低温响应过程中发挥重要作用。Sperotto等[61]对处于营养生长期的两种基因型水稻进行了低温(10 ℃)诱导下的转录组分析,在耐低温和低温敏感型水稻中分别获得了831和357个差异表达基因,并发现与细胞壁组成相关的基因在耐低温植株中大量表达,使低温下植株中有较高的纤维素积累;与脂类代谢相关的基因也在耐低温植株中大量表达,使其积累了较多的亚油酸。且相比于敏感型,耐低温植株具有较高的光合效率和抗氧化能力,以及更有效的乙烯、Ca2+信号通路。Chen等[62]对高温处理下的二穗短柄草(Brachypodiumdistachyon)幼苗叶片,以及抽穗期的叶片和花序进行了RNA-seq分析,656个基因在3组样品中均发生了差异表达,差异基因大多与光合天线蛋白、内质网及剪接体相关;还发现1973个可变剪接。综上所述,通过转录组测序能够发现生长调节因子Karrikins、转录因子(CBF2、AP2/ERF、NAC、MYB、bZIP和CBF3)、亚油酸、纤维素、Ca2+和乙烯信号通路以及细胞壁组成相关基因在植物应对低温胁迫过程中发挥重要功能。
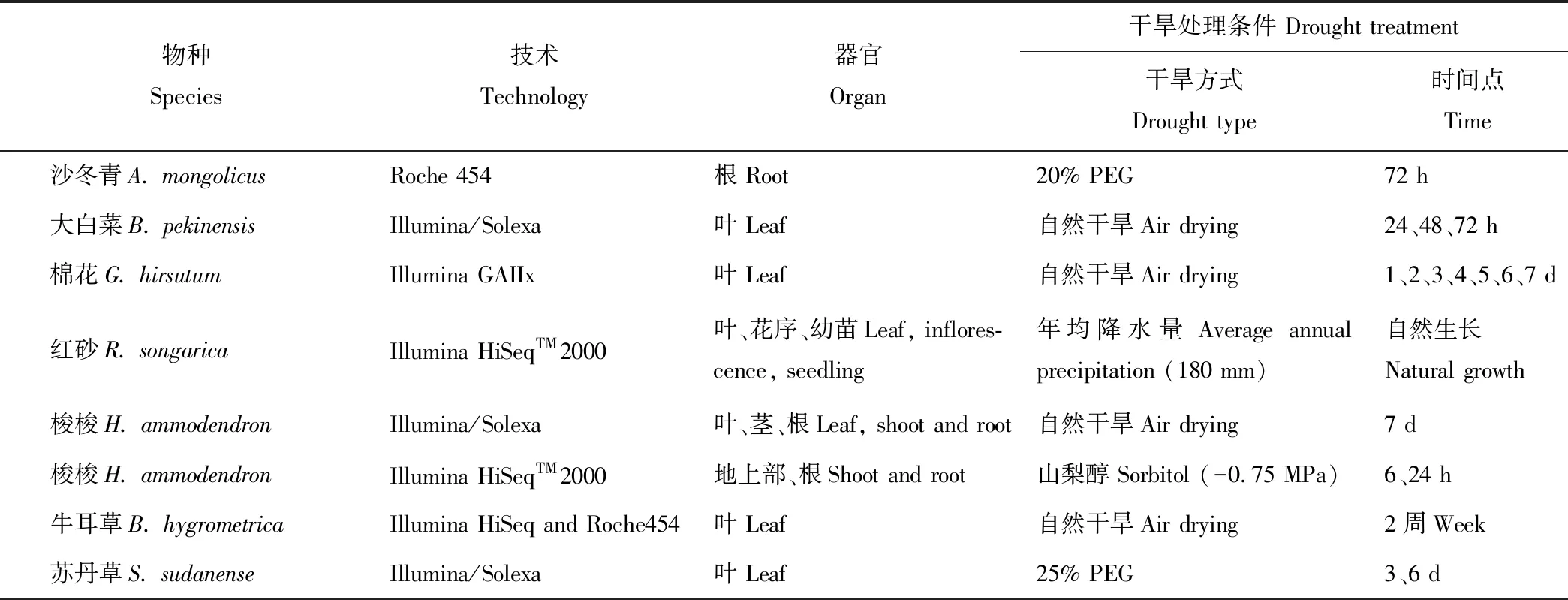
表3 部分植物耐旱分子机制的转录组学研究
2.4 养分胁迫下林草植物转录组学研究
排根的形成对于植物适应磷饥饿具有重要作用,羽扇豆(Lupinusmicranthus)因为有发育良好的排根,能够在低磷条件下正常生长。O’Rourke等[63]利用RNA-seq技术分析缺磷状态下羽扇豆叶片、根以及排根的基因表达及其适应机理。通过测序组装获得了平均长度为1155 bp的125821个Unigenes,其中50734个基因具有转录活性。基因表达分析总共检测到2128个差异表达基因,叶片中有1342个(987个在磷充足叶片上调),根中有904个(396个在磷充足根中上调),包括110个磷酸盐转运蛋白和155个转录因子,有12个基因在拟南芥和马铃薯中也存在差异表达。之后,Secco等[64]利用Solexa测序技术研究磷饥饿下羽扇豆排根的发育和调控机理,通过测序组装,产生了46383个contigs,再并入到已公布的羽扇豆转录组数据集,最后形成高质量的65097个contigs。通过功能注释,磷饥饿下在排根发育的不同阶段发现许多差异表达基因,成熟的排根中有许多与膜转运蛋白和代谢适应相关的基因表达,根尖中只有直接参与磷吸收与平衡的基因有表达,而与分生组织活性和发育进程相关的转录因子以及细胞分裂素和生长素基因在排根尖中有较高表达,说明转录因子和激素在排根发育过程中发挥重要作用,而且不同生长阶段的排根在耐磷饥饿过程中功能不尽相同。邵彩虹等[65]利用RNA-seq技术对水稻根系响应氮素胁迫的分子机制进行了分析,发现胁迫下,根系有2270个基因发生了差异表达,其中木质素合成关键酶肉桂酰辅酶A还原酶基因、参与细胞伸长发育的EGase基因以及与生长素和新RNA合成相关的基因上调表达,以促进根系伸长生长来应对养分胁迫刺激。
通过RNA测序,可以分析植物在高盐、干旱、低温、高温及营养匮缺等非生物胁迫下的基因表达及信号通路变化,从而筛选出一些胁迫响应基因;也可以通过比较两个不同抗性品种在非生物胁迫前后的基因表达差异,直接鉴定出抗逆关键基因,探究植物适应机理,对关键基因分子功能进行更深一步的研究,并应用于牧草及作物的抗逆遗传改良。
3 转录组学与其他功能基因组学相结合研究
RNA-seq与DNA测序相结合可以检测SNP、RNA编辑以及表达数量性状位点,尤其在分析中将基因型数据和基因表达变化进行相关分析可以获得复杂性状的遗传机理,例如植株高度,且表达数量性状位点研究发现遗传变异能够影响许多基因的表达[66-67]。RNA-seq与重测序结合可以在融合基因分析中去除假阳性,还能分析拷贝数的改变[68-69]。通过将RNA-seq与DNA甲基化进行整合,对差异表达基因和甲基化模式进行关联分析,能够识别具有差异表达和差异甲基化的一个或多个基因[70]。RNA-seq与转录因子染色质免疫共沉淀测序(chromatin immunoprecipitation, ChIP)结合分析可以去除ChIP-seq中的假阳性,并能检测出转录因子对其目标基因的激活或抑制效应。RNA-seq与miRNA-seq整合分析能够阐明miRNAs对转录稳态水平的调节效应。RNA-seq与蛋白质组学分析相关性不高,但二者整合分析能够鉴定新的亚型,RNA-seq结果中没有检测到某个基因的表达,但在质谱分析中发现它的多肽,说明发生了翻译后修饰。RNA-seq与代谢组学相结合能够鉴定出在基因表达和代谢水平均发生了变化的信号通路。另外,还能通过一些软件或在线网站将多个基因组数据进行整合分析。SNMNMF和PIMiM能够将mRNA和 miRNA表达数据与蛋白-蛋白、DNA-蛋白、miRNA-mRNA互作网络结合起来鉴定miRNA-基因调控模式[71-72]。MONA能够将不同水平的功能基因组学数据结合起来,包括mRNA、miRNA、DNA甲基化和蛋白质组学,以检测样品生物学功能的改变[73]。Paintomics可以整合任何类型的基因组学数据进行通路分析[74]。3Omics可以对转录组、代谢组和蛋白质组进行关联分析,研究其调控网络[75]。目前需要做的就是要把转录组数据与其他组学相结合,从多方面更系统、更深入地对数据进行分析,获得更充分、更精确的研究结果。
4 展望
新一代高通量测序技术已经广泛应用于林草植物抗逆性研究中,并且取得了较好的成果。但是RNA-seq数据结果容易受参数设置影响,尤其是低丰度表达基因。相信随着测序技术的不断发展,即测序时间和成本的不断压缩,以及测序通量和准确度的不断提高,用少量的mRNA就能构建cDNA文库用于测序,并获得较长的reads,得到优质的且精确度较高的转录本;在接下来的几年中,RNA-seq分析会越来越完善,也必定会带来更加惊人的发现。此外,以单分子测序仪,单分子实时测序(single molecule real time,SMRT)以及即时DNA测序技术为主的第三代测序技术,即单分子测序逐渐呈现,它不需要进行PCR扩增,可直接对DNA分子全长进行单独测序。但是三代测序技术刚起步,还不够完善、成熟,每次对单个细胞只能进行一种功能基因组的检测,不能在检测转录组的同时进行其他组学分析,且碱基错误率较高,费用昂贵,未能得到大面积使用。今后的发展趋势是要不断改进并创造性的综合利用各种测序技术,将RNA-seq与三代测序技术相结合来进行纠错,降低错误率,获得丰富且精确的转录本。还要将转录组学与蛋白组学以及代谢组学等其他组学相关联,进行整合分析,更好地应用于植物抗逆关键基因的鉴定及抗逆分子机理研究。
猜你喜欢
中国生殖健康(2020年4期)2021-01-18
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
天然产物研究与开发(2018年2期)2018-04-04
红领巾·萌芽(2017年5期)2017-06-23
爆笑show(2016年7期)2017-02-09
少儿科学周刊·儿童版(2015年10期)2015-11-07
少儿科学周刊·儿童版(2015年1期)2015-07-07
医学研究杂志(2015年11期)2015-06-10
