
非平面四吡咯锌配合物的吸收光谱特征
2019-12-19 02:05王鑫
科技创新与应用 2019年35期
王鑫
摘 要:四吡咯锌配合物可有几种不同的几何结构,如锌卟啉配合物、四联吡咯锌配合物、三联吡咯锌配合物、二联吡咯锌配合物和单吡咯锌配合物。研究π共轭效应对此系列四吡咯锌配合物吸收光谱性质的影响具有重要的学术意义,且可为新型四吡咯锌类材料的设计方面提供有价值的参考。
关键词:四吡咯锌卟啉配合物;π共轭结构;吸收光谱性质
中图分类号:O641 文献标志码:A 文章编号:2095-2945(2019)35-0053-02
Abstract: Tetrapyrrole zinc complexes can have several different geometric structures, such as zinc porphyrin complexes, tetrapyrrole zinc complexes, tripyrrole zinc complexes, bipyrrole zinc complexes and monopyrrole zinc complexes. The study of π conjugation effect on the absorption spectral properties of this series of tetrapyrrole zinc complexes is of great academic significance, and can provide a valuable reference for the design of new tetrapyrrole zinc materials.
Keywords: tetrapyrrole zinc porphyrin complex; π conjugated structure; absorption spectral properties
引言
锌卟啉是最为典型的四吡咯锌配合物,具有特殊的电子结构及优良的光电性质[1-3]。并且,通过修饰可有效调节的锌卟啉衍生物的光电化学性质,使其应用领域及应用价值得到进一步的拓展[4-6]。目前,人们对锌卟啉配合物的修饰主要是在保持锌卟啉的平面结构的基础上将不同数目及不同功能的官能团引入其外围[6-8]。
相比于具有平面结构的锌卟啉衍生物,具有非平面结构的锌卟啉衍生物的光电化学性质也引起了研究者的兴趣[1,2,9,10]。非平面锌卟啉衍生物的合成方法多种多样,如:利用空间位阻效应,将大体积的取代基引入锌卟啉配合物外围或轴向位置使其扭曲;通过降低刚性,变换锌卟啉配合物中的次甲基桥键使其扭曲。将锌卟啉配合物中的次甲基桥键逐渐破坏,可以得到一系列具有不同π共轭结构的非平面四吡咯锌衍生物,如四联吡咯锌配合物、三联吡咯锌配合物、二联吡咯锌配合物及单吡咯锌配合物。本文综述了此系列化合物的前线分子轨道特征及吸收光谱特征,希望能为新型、高效的四吡咯锌类材料的设计提供理论参考。
1 几何结构和前线分子轨道性质
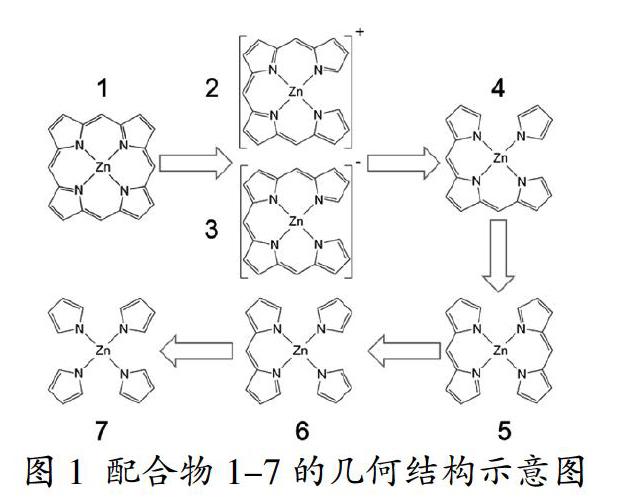
在此,配合物1代表锌卟啉配合物。当锌卟啉配合物1的一个次甲基桥键被破坏后,可得到正一价的四联吡咯锌配合物2和负一价的四联吡咯配合物3;当锌卟啉配合物1的两个次甲基桥键被破坏后,可得到三联吡咯配合物4和二联吡咯配合物5;当锌卟啉配合物1的三个次甲基桥键被破坏后,可得到二联吡咯配合物6;当锌卟啉配合物1的四个次甲基桥键被破坏后,可得单吡咯锌配合物7,如图1所示。
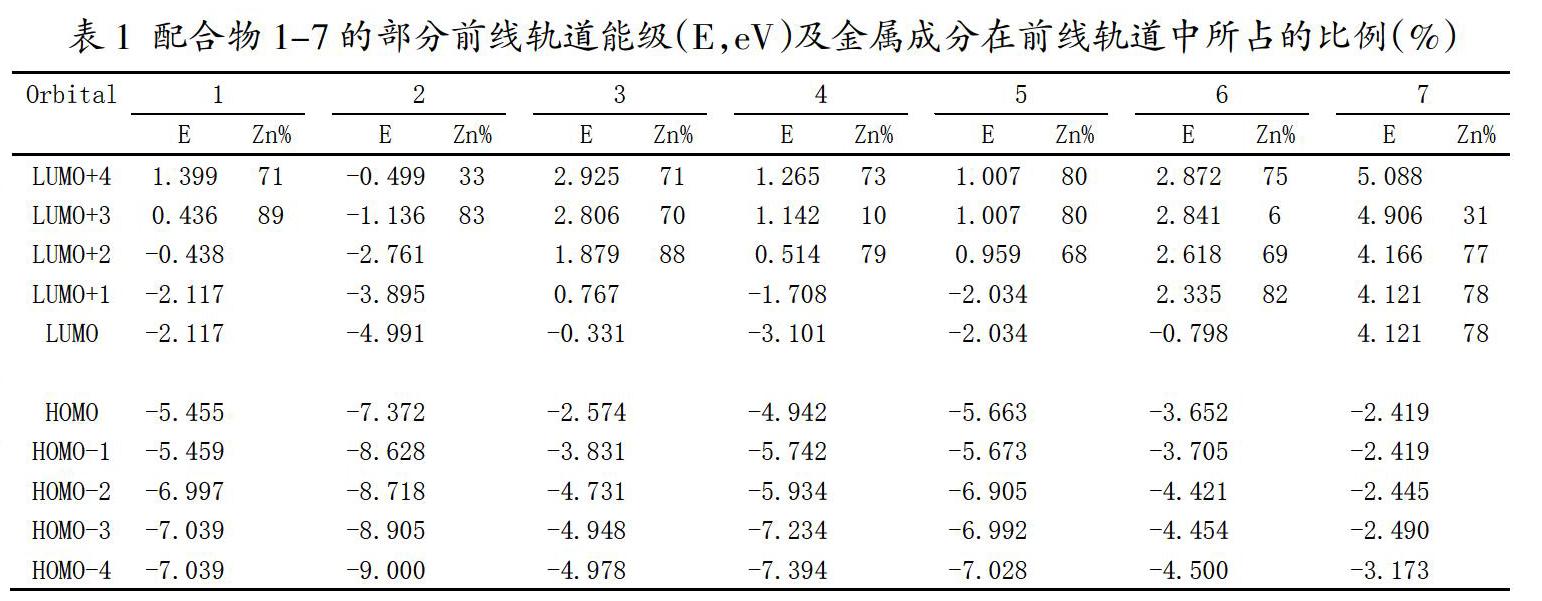
通过表1我们可以看出锌卟啉配合物1的两个最高的占據轨道几乎是简并的,两个最低的未占据轨道是完全简并的,并且这四条轨道与其它前线分子轨道在能量上有明显分离。这也是经典的Gouterman四轨道模型理论中所描述的[11,12]。但是,对于四联吡咯锌配合物2和3、三联吡咯锌配合物4和二联吡咯锌配合物6,其两个最高占据轨道及两个最低未占据轨道在能量上不再简并,并且,相对于锌卟啉配合物1,配合物2、3、4及6的这四条前线分子轨道与其它分子轨道在能量上不再有明显分离。此外,随着π共轭骨架的打破,配合物1-7的未占据轨道中金属成分越来越多。总之,当锌卟啉的π共轭骨架被逐渐破坏后,其前线分子轨道的分布和组成都发生了显著的变化。
2 电子吸收光谱
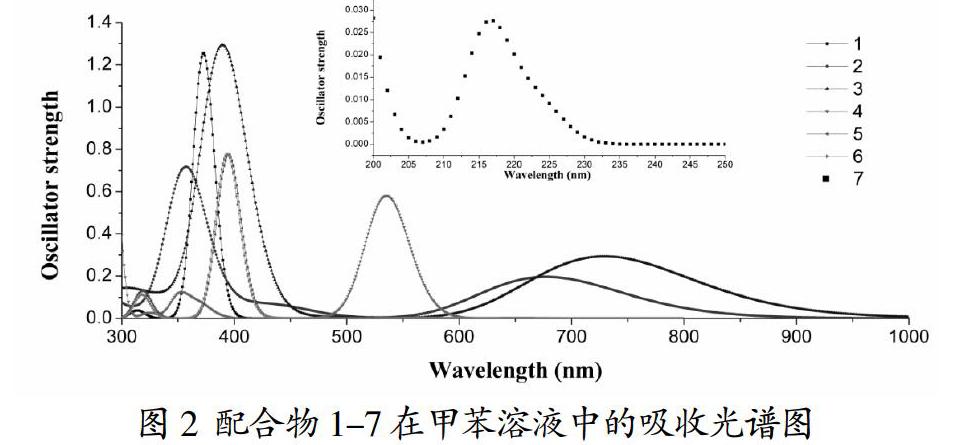
锌卟啉及其衍生物的吸收光谱特性一直都在实验和理论研究中备受关注[13-16]。在卟啉类化合物的吸收光谱中,我们把420nm处左右的强吸收称为B带吸收,把500~750nm处的弱吸收称为Q带吸收。从图2中我们可以看出,锌卟啉配合物1有一个较弱的Q带吸收和一个较强的B带吸收,为典型的卟啉吸收光谱特征。但是,当锌卟啉的π共轭骨架被逐渐破坏后,配合物2-7的吸收波长和吸收强度都发生了很大的变化。
从图2中我们可以明显的看到,相比于配合物1的B带和Q带吸收,配合物2-4在B带吸收明显减弱,但是其在Q带吸收强度却大大增强。所以,相比于锌卟啉配合物,四联吡咯锌配合物和三联吡咯锌配合物在可见光区具有较强光捕获能力。然而,二联吡咯和单吡咯锌配合物5-7的Q带吸收就并没有增强,尤其是在单吡咯配合物7中,已经没有卟啉类化合物的B带和Q带吸收,只在220nm左右有一个极其微弱的吸收峰。因此,π共轭效应对四吡咯锌配合物的吸收光谱特征产生了显著的影响。
3 结论
本文以非平面四吡咯锌配合物为模型,综述了π共轭效应对四吡咯锌配合物的前线分子轨道及吸收光谱性质的影响。当锌卟啉配合物的π共轭骨架被破坏后,其两个最高占据轨道和两个最低未占据轨道不再简并,并且与其它前线分子轨道在能量上越来越接近;其未占据轨道中的金属成分所占的比例也越来越大;其Q带吸收强度呈现出先增强后减弱的特征;其B带吸收强度则表现出逐渐减弱的特征。希望通过探讨此系列吡咯锌配合物前线分子轨道特征和吸收光谱特征,能够为设计高效可控的四吡咯锌配合物材料提供依据。
參考文献:
[1]WANG X, BAI F-Q, XIE M, et al. A theoretical investigation on the π-conjugation effect on the structures and spectral properties of tetra pyrrole zinc complexes [J]. Synthetic Metals, 2015, 210 Part B 258-267.
[2]WANG X, BAI F-Q, LIU Y-T, et al. Theoretical investigation on the spectroscopic properties of Zn porphyrin and Zn tetrapyrrin [J]. Synthetic Metals, 2016, 21318-24.
[3]ROCHFORD J, GALOPPINI E. Zinc (II) tetraarylporphyrins anchored to TiO2, ZnO, and ZrO2 nanoparticle films through rigid-rod linkers[J]. Langmuir, 2008,24(10):5366-5374.
[4]SEO K D, LEE M J, SONG H M, et al. Novel D-π-A system based on zinc porphyrin dyes for dye-sensitized solar cells: Synthesis, electrochemical, and photovoltaic properties [J]. Dyes and Pigments, 2012,94(1):143-149.
[5]VIOLA E, DONZELLO M P, SCISCIONE F, et al. Tetra-2,3-pyrazinoporphyrazines with externally appended pyridine rings. 17. Photosensitizing properties and cellular effects of ZnII octacationic and ZnII/PtII hexacationic macrocycles in aqueous media: Perspectives of multimodal anticancer potentialities [J]. Journal of Photochemistry and Photobiology B: Biology, 2017,169101-109.
[6]XIA H-Q, CHEN J, BAI F-Q, et al. Computational study on zinc porphyrin analogs for use in dye-sensitized solar cells [J]. Journal of Porphyrins and Phthalocyanines, 2014,18(05):406-415.
[7]WALSH P J, GORDON K C, OFFICER D L, et al. A DFT study of the optical properties of substituted Zn (II) TPP complexes[J]. Journal of Molecular Structure: THEOCHEM, 2006,759(1-3):17-24.
[8]GAO Y, GUAN W, YAN L, et al. Theoretical screening of promising donor and π-linker groups for POM-based Zn-porphyrin dyes in dye-sensitized solar cells [J]. Physical Chemistry Chemical Physics, 2019,21(7):3822-3831.
[9]BALLICO M, RAPOZZI V, XODO L E, et al. Metallation of pentaphyrin with Lu(III) dramatically increases reactive-oxygen species production and cell phototoxicity [J]. European journal of medicinal chemistry, 2011,46(2):712-720.
[10]WANG X, BAI F-Q, LIU Y, et al. A Computational Way To Achieve More Effective Candidates for Photodynamic Therapy [J]. Journal of Chemical Information and Modeling, 2017,57(5):1089-1100.
[11]GOUTERMAN M. Study of the Effects of Substitution on the Absorption Spectra of Porphin [J]. The Journal of Chemical Physics, 1959,30(5):1139-1161.
[12]GOUTERMAN M. Spectra of porphyrins [J]. Journal of Molecular Spectroscopy, 1961,6138-163.
[13]HASHIMOTO T, CHOE Y-K, NAKANO H, et al. Theoretical Study of the Q and B Bands of Free-Base, Magnesium, and Zinc Porphyrins, and Their Derivatives [J]. The Journal of Physical Chemistry A, 1999,103(12): 1894-1904.
[14]KERRIDGE A. A RASSCF study of free base, magnesium and zinc porphyrins: accuracy versus efficiency [J]. Physical Chemistry Chemical Physics, 2013,15(6):2197-2209.
[15]MINAEV B, WANG Y-H, WANG C-K, et al. Density functional theory study of vibronic structure of the first absorption Q x band in free-base porphin [J]. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2006,65(2):308-323.
[16]NAKAI K, SAHNOUN R, KATO T, et al. Time-dependent density functional theory investigation of electric field effects on absorption spectra of meso-meso-linked zinc porphyrin arrays: Role of charge-transfer states [J]. The Journal of Physical Chemistry B, 2005,109(29):13921-13927.
