
微RNA-29b在心肌缺血再灌注损伤中的作用及机制研究
2019-12-11 03:49高英英
安徽医药 2019年12期
高英英
急性心肌梗死是冠状动脉急性、持续性缺血缺氧所引起的心肌坏死[1]。近年来,随着各地区胸痛中心的建立以及溶栓治疗、支架植入术的进步,急性心梗病人的病死率明显降低[2]。但是,由于尚无有效改善冠状动脉血管再灌注损伤的治疗方案,急性心梗病人的预后受到影响。因此,本实验探究小分子RNA在心肌缺血再灌注损伤中的作用,进一步阐明心肌缺血再灌注损伤的病理过程,为临床治疗心肌缺血再灌注损伤提供思路。
既往研究表明组织缺血再灌注损伤的潜在机制主要包括自由基损伤,钙超载,白细胞激活和微血管损伤等病理过程[3-5]。这些病理过程会造成大量心肌细胞的凋亡和坏死,但是也为临床治疗提供新的靶点。因此,深入研究心肌缺血再灌注损伤的病理过程对减少急性心肌梗死病人PCI植入术后再灌注损伤具有重要价值。
微RNA(miRNA)是短的单链RNA序列(通常为19~23个核苷酸),广泛表达于生物系统中,其作用为控制基因在多种生理和病理过程中的表达,因此在转录后调控中具有关键作用[6-7]。Huang Z等[8]的研究结果表明miR-29b通过作用于位于1q21区域的myeloid cell leukemia-1(MCL-1)蛋白从而调控脑缺血再灌注损伤导致的神经细胞凋亡,然而尚没有文献报道其在心肌缺血再灌注损伤中的作用,因此在本实验中我们检测了miR-29b在心肌缺血/再灌注(Ischemia/Reperfusion,I/R)损伤时的表达变化,同时通过上调其表达水平明确探讨其在心肌缺血再灌注损伤中的作用。
本研究于2018年2月至2019年2月成功构建了缺血再灌注细胞模型以及心肌缺血再灌注动物模型,通过上调miR-29b表达,观察miR-29b能否影响缺血再灌注时心肌细胞的凋亡水平,并进一步明确miR-29b在缺血再灌注损伤中发挥心脏保护作用的具体信号通路,从而阐明miR-29b在心肌缺血再灌注损伤中的作用及机制。
1 材料与方法
1.1 细胞与动物H9C2心肌细胞系购于上海生化细胞所,于37℃、5%二氧化碳、饱和湿度培养箱中培养,用含有10%胎牛血清的DMEM培养液培养。30只健康清洁级雄性C57/B6小鼠,6~7周龄,体质量范围为16~18 g,购于上海斯莱克实验动物有限公司,动物生产许可证编号:SCXK(沪)2012-0002。
1.2 主要试剂miR-29b类似物(mimic)、NC miRNA、miR-29b激动剂(agomir)购于广州锐博生物有限公司,叔丁基过氧化氢(TBHP)购于美国sigma公司,兔抗小鼠BCL2-Associated X的蛋白质(Bax)、B淋巴细胞瘤-2(BCL-2)单克隆抗体均购于Abcam公司;兔抗小鼠磷酸化蛋白激酶B(p-Akt)、蛋白激酶B(Akt)单克隆抗购于CST公司;甘油醛-3-磷酸脱氢酶(GAPDH)单克隆抗体、辣根过氧化物酶标记羊抗兔IgG均购于Bioworld公司;人胆囊收缩素/缩胆囊素八肽(CCK-8)购于伯信生物公司;2,3,5-氯化三苯基四氮唑(TTC)染色试剂购于Solarbio公司;总RNA提取试剂盒(东洋纺)以及miR-29b、GAPDH引物购于伯信生物公司;实时荧光定量多聚核苷酸链式反应(qPCR)仪购于Eppendorf公司。
1.3 主要方法通过100 μmol/L TBHP诱导H9C2心肌细胞损伤构建I/R细胞模型;分别设立正常对照组,TBHP处理组,NC miRNA处理组,miR-29b mimic处理组。通过qPCR检测各组心肌细胞内miR-29b表达量;CCK-8检测各组心肌细胞的存活率;蛋白质印迹法(Western Blot)检测心肌细胞中凋亡蛋白和蛋白激酶B(Akt)蛋白磷酸化水平的改变。另一方面,将30只C57/B6小鼠(6~7周龄)逐个编号,按随机数字表法分为三组:I/R组、NC miRNA处理组和miR-29b agomir处理组,其中miR-29b agomir组小鼠预先给予miR-29b agomir治疗。结扎冠状动脉左前降支30 min,然后再灌注4 h建立心肌缺血再灌注动物模型。通过埃文斯蓝(Evans)和TTC双重染色评估三组小鼠心肌梗死面积;qPCR检测各组小鼠心肌组织内miR-29b表达量;蛋白质印迹法检测各组小鼠心肌组织内凋亡蛋白改变。
1.3.1 细胞活性测定 将H9C2心肌细胞以每孔5 000个细胞浓度接种于96孔板中,然后暴露于不同浓度的TBHP12 h后,通过CCK-8测定细胞存活率,选择最为合适的TBHP浓度用于心肌细胞模型构建。另一方面,通过上调miR-29b表达量明确其对于心肌细胞活性的影响。CCK-8的具体方法为:将CCK-8染料加入每个孔中并孵育3 h。通过评估490 nm处的吸光度来测量存活细胞数。为了实验结果的一致性,将CCK-8测定重复3次。
1.3.2 细胞及动物模型构建 通过CCK-8摸索适宜浓度TBHP构建缺血再灌注细胞模型。通过吸入异氟烷麻醉C57/B6小鼠,暴露气管,小动物呼吸机辅助呼吸,从左侧第4肋间进入胸腔,用7-0滑线结扎冠状动脉左前降支30 min,然后解开结扎线,冠状动脉再灌注4 h,建立心肌缺血再灌注动物模型。再灌注4 h后处死各组小鼠,部分心脏储存于-80℃冰柜内,部分心脏用于TTC染色。
1.3.3 qPCR检测mRNA表达量 取出放置于-80℃保存的各组心肌组织,提取心肌组织中总RNA并测定总RNA浓度及纯度,按东洋纺逆转录试剂盒说明书操作合成cDNA,通过Eppendorf MAS荧光定量PCR分析系统进行检测,以U6为参照,目的基因的相对含量以2-ΔΔCt表示,引物序列见表1。

表1 PCR引物序列
1.3.4 蛋白质印迹法检测p-Akt、Akt、Bax和BCL-2的蛋白水平 通过细胞裂解液降解心肌组织及心肌细胞蛋白,离心后取上清测定蛋白含量,经凝胶电泳分离和转膜,5%脱脂奶粉室温封闭2 h,TBST洗膜后分别加一抗GAPDH(1∶5 000)、p-Akt(1∶1 000)、Akt(1∶1 000)、Bax(1∶1 000)和 BCL-2(1∶1 000),4℃孵育过夜,二抗室温孵育2 h,含吐温的三乙醇胺缓冲盐水溶液(tris buffered saline+tween 20,TBST)洗膜3次后用化学发光试剂(electrochemiluminescence,ECL)显影曝光。Bax和BCL-2以GAPDH蛋白条带作为参照,磷酸化的Akt以总Akt蛋白条带作为参照,通过Bio-Rad凝胶成像系统扫描,Image Lab图像软件定量分析每个条带灰度值。
1.3.5 心肌梗死面积计算 通过Evans和TTC双重染色方法评估心肌损伤范围。在心肌再灌注结束时,将0.2 mL 2%Evans蓝染料注入下腔静脉。当心脏右侧变蓝时,迅速取出心脏,用0.9%氯化钠溶液冲洗,并在-20℃冷冻。制备5个1 mm厚的心脏切片,并在1%TTC染料中37℃温育20 min。TTC染成淡红色的活组织被定义为危险区(AAR);伊文思蓝染色的非缺血性心肌呈深蓝色;梗死区(INF)在染色后显苍白色。评估同一切面各组小鼠心肌组织梗死面积大小。
1.4 统计学方法用SPSS 22.0统计软件进行分析。数据采用xˉ±s表示,多组间比较采用单因素方差分析(one-way ANOVA),组间两两比较采用Bonferroni校正的t检验,以P<0.05为差异有统计学意义。
2 结果
2.1 心肌细胞内miR-29b的表达水平在本研究中,我们通过qPCR检测转染不同类型小分子RNA后心肌细胞内miR-29b的表达水平。实验结果提示转染miR-29b mimic后,miR-29b的表达水平(450.83±36.88)显著增加,且较NC miRNA组(1.43±0.10)、正常对照组(1.29±0.20)明显上调(均P<0.05)。
2.2 miR-29b表达水平对心肌细胞活性的影响CCK-8 实验结果表明,100 μmol/L 和 200 μmol/L TBHP组能够显著抑制心肌细胞活性(P<0.05)结果分别为(57±7)%,(52±4)%。给予 miR-29b mimic治疗后心肌细胞存活率(85±3)%较NC miRNA(64±3)%及TBHP细胞模型组(65±3)%明显上升(P<0.05)。CCK-8实验结果表明,上调miR-29b表达可以增加缺血再灌注损伤后心肌细胞的存活率。
2.3 上调miR-29b可降低心肌细胞内凋亡蛋白表达蛋白质印迹法结果提示,与100 μmol/L TBHP处理组Bax/BCL-2的比值(4.37±0.37)相比,miR-29b mimic处理组心肌细胞内Bax蛋白表达水平明显降低,BCL-2蛋白表达水平明显升高(见图1A),Bax/BCL-2的比值(0.86±0.07)明显的下调(P<0.05,见图1B)。NC miRNA处理组心肌细胞凋亡蛋白Bax/BCL-2的比值(4.26±0.41)较100 μmol/L TBHP处理组无明显改变(P>0.05,见图1B)。这些实验结果表明miR-29b可以显著降低心肌细胞凋亡水平,从而保护心肌细胞。
2.4 上调miR-29b表达可能通过Akt信号通路调控凋亡通过检测各组心肌细胞中p-Akt、Akt蛋白表达水平明确miR-29b在心肌细胞缺血再灌注损伤中的具体调控机制。蛋白质印迹法结果提示TBHP处理后心肌细胞内Akt磷酸化水平(0.81±0.05)较正常对照组(0.22±0.01)明显增高(P<0.05)。上调miR-29b表达后,miR-29b mimic组Akt蛋白磷酸化水平(0.41±0.02)较TBHP处理组明显降低(P<0.05)。这些实验结果表明miR-29b可能通过下调Akt蛋白磷酸化水平,发挥缺血再灌注损伤时的心脏保护作用。见图2。
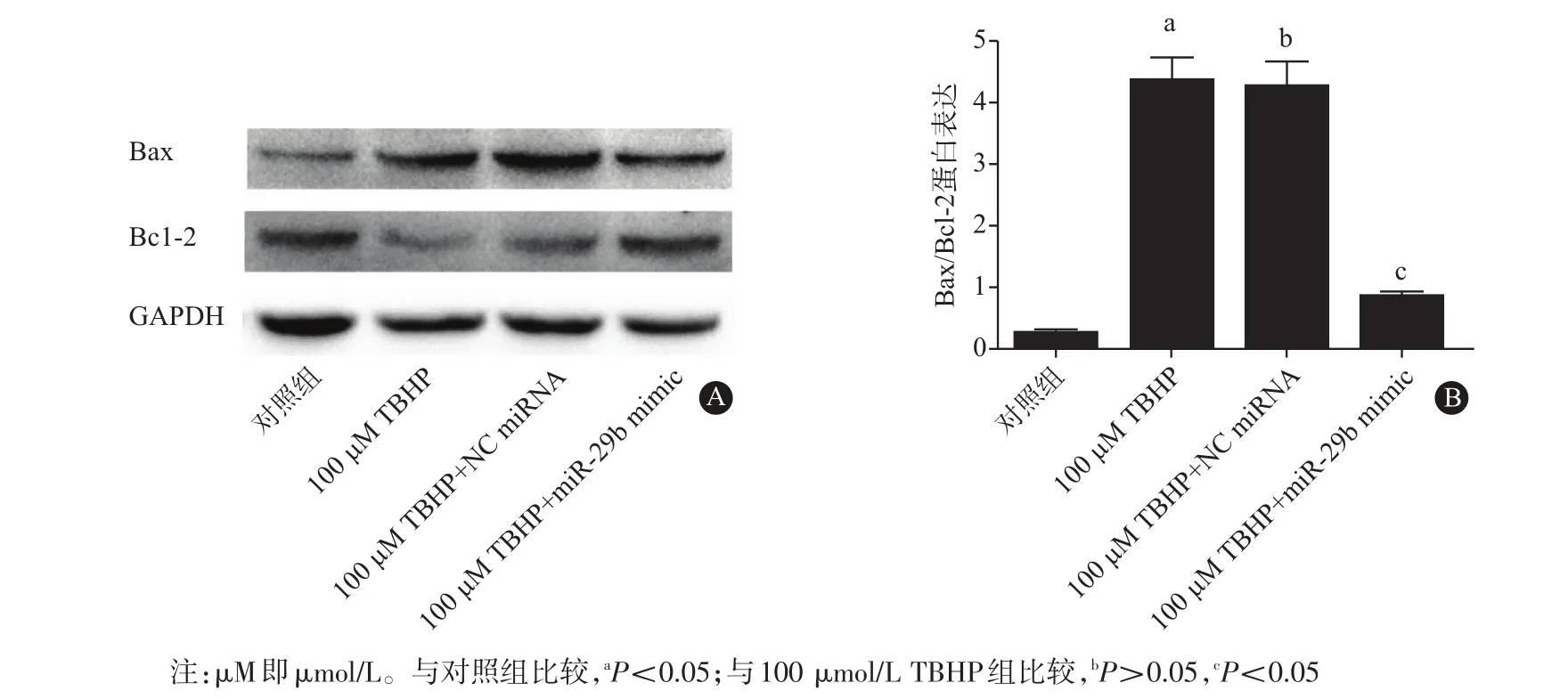
图1 miR-29b对于100 μmol/L叔丁基过氧化氢(TBHP)处理后心肌细胞凋亡蛋白Bax水平的影响:A为蛋白电泳图;B为量化图

图2 miR-29b对于100 μmol/L叔丁基过氧化氢(TBHP)处理后心肌细胞Akt蛋白磷酸化水平的影响:A为蛋白电泳图;B为量化图
2.5 上调miR-29b表达可改善心肌梗死面积Evans蓝和TTC双重染色结果表明,I/R组小鼠(62±2)%和NC miRNA处理组小鼠心肌梗死范围(60±4)%无明显改变(P>0.05),而给予miR-29b agomir处理(24±3)%后可有效减少缺血再灌注导致的心肌梗死范围,增加心肌细胞存活率(P<0.05)。见图3。
2.6 上调miR-29b表达可减少心肌组织内凋亡相关蛋白的表达我们在动物模型中再次验证miR-29b对于心肌组织凋亡的保护作用。蛋白质印迹法结果提示,与I/R组小鼠Bax/BCL-2的比值(3.31±0.24)和NC miRNA处理组小鼠Bax/BCL-2的比值(3.24±0.32)相比,miR-29b agomir处理组小鼠心肌组织内Bax蛋白表达明显降低,BCL-2蛋白表达水平明显升高,因此,miR-29b agomir处理组小鼠心肌组织内Bax/BCL-2的蛋白比值(0.32±0.04)明显下调(P<0.05,见图4),该结果表明上调miR-29b表达水平可以显著降低缺血再灌注后心肌组织的凋亡水平。
3 讨论
急性心肌梗死是心血管危重症,病死率高,危害性大,随着医疗技术的进步,我们已能够在心肌梗死发病的早期就对其进行干预,临床上的治疗方法包括:药物溶栓、支架介入和外科搭桥手术[9]。但是对于心肌缺血再灌注损伤的机制尚未完全阐明,影响临床上急性心肌梗死病人的预后。miRNA是一类参与转录后基因表达调控的小分子RNA,已被证明参与机体多种病理生理过程。因此,在本实验中我们通过转染技术调控心肌缺血再灌注细胞及动物模型中miR-29b表达水平,并进一步检测心肌细胞凋亡水平以及信号通路改变,明确miR-29b在心肌缺血再灌注损伤中的作用及其潜在机制。
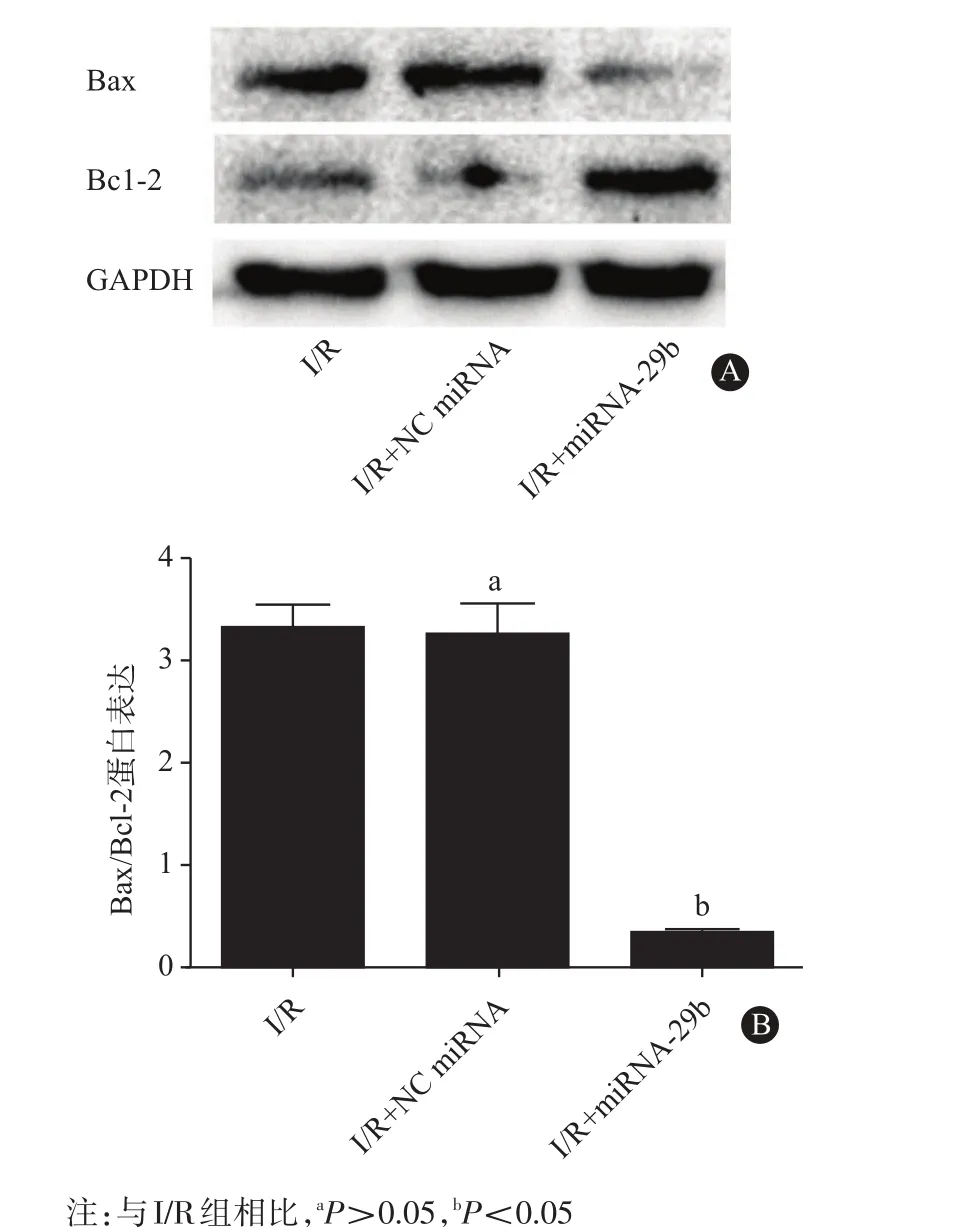
图4 miR-29b激动剂(agomir)治疗对于心肌缺血再灌注损伤小鼠心肌组织凋亡蛋白表达水平的影响:A为蛋白电泳图;B为量化图
既往已有大量研究表明miR-29b在不同疾病中发挥重要的作用。有研究发现miR-29b可以靶向作用于Sp1蛋白,然后通过抑癌基因PTEN/Akt信号通路抑制舌鳞状细胞癌中癌细胞的增殖,迁移和侵袭[10]。另有研究结果表明miR-29b通过靶向磷脂酰肌醇-3激酶(PI3K)/Akt信号传导途径调节血管紧张素Ⅱ诱导的高血压大鼠肾纤维化过程[11];Wu Y等[12]的研究表明 miR-1 和 miR-29b 可通过抑制PI3K/Akt信号通路,对心肌梗死后心室重构产生不良影响。因此我们猜想miR-29b在心肌缺血再灌注中发挥重要作用,且Akt信号通路是其发挥作用的主要的信号通路。
在本实验中我们发现上调miR-29b表达能够有效减少心肌组织以及心肌细胞中凋亡蛋白Bax的表达,同时提高BCL-2蛋白的表达,这一结果表明miR-29b在心肌缺血再灌注损伤中发挥抑制心肌细胞凋亡的作用。进一步的实验表明,上调miR-29b表达能够下调Akt蛋白磷酸化水平,因此我们推测miR-29b可能通过调控Akt信号通路改善心肌缺血再灌注时心肌细胞的凋亡,从而发挥心脏保护作用。
本实验的最大局限之处是没有设立Akt信号通路激动剂处理组,因而尚不能说明一定是Akt信号通路在miR-29b抗凋亡中发挥作用。因此对于miR-29b通过Akt信号通路调控心肌细胞凋亡这一结论还有待进一步研究。
综上所述,本研究给予心肌缺血再灌注模型miR-29b处理,发现miR-29b组较I/R组的心肌细胞存活率、梗死面积均有明显改善,心肌细胞凋亡水平降低,并且发现miR-29b能够降低Akt的蛋白磷酸化水平,表明miR-29b可能通过该通路减少缺血再灌注时的心肌细胞凋亡,从而保护心脏。
(本文图3见插图12-2)

图3 miR-29b激动剂(agomir)治疗改善心肌缺血再灌注造成的心肌组织损伤:A为染色图(Evans蓝和TTC双重染色);B为量化图
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年7期)2021-11-04
天津医科大学学报(2019年6期)2019-08-13
分析化学(2017年12期)2017-12-25
海南医学(2016年8期)2016-06-08
中国病理生理杂志(2015年8期)2015-12-21
安徽医科大学学报(2015年9期)2015-12-16
遗传(2014年3期)2014-02-28
现代农业科技(2009年19期)2009-03-20
中学生数理化·八年级数学华师大版(2008年3期)2008-08-26
