
从遗传力到肠菌力:概念及研究进展
2019-11-28 12:01文超良孙从佼杨宁
遗传 2019年11期
文超良,孙从佼,杨宁
从遗传力到肠菌力:概念及研究进展
文超良,孙从佼,杨宁
中国农业大学动物科技学院,畜禽国家育种工程实验室,北京 100193
遗传力是数量遗传学中衡量性状遗传变异的重要参数之一,对动植物遗传育种、医学和进化生物学均具有重要意义。自1918年Fisher提出方差组分剖分思想后,遗传力的分析模型和估计方法不断迭代更新。近年来,一些研究者在研究肠道微生物与宿主表型关系的热点问题中,引入类似于遗传力的概念和估计方法,系统评估动物机体性状表现受肠道微生物因素的影响程度,本文将这一参数(microbiability,2)译为“肠菌力”。文章简要介绍了遗传力估计的发展历程,概述了肠道菌群与宿主基因组的复杂关系,重点阐释了肠菌力的概念和估计方法,以期为肠道微生物组塑造宿主表型变异的研究提供一定的借鉴和参考。
遗传力;肠菌力;关联分析;数量性状;方差组分;关系矩阵
动物是由宿主机体和共栖微生物构成的“共生总体(Holobiont)”[1]。宿主性状的表现既受其自身基因的调控,又受到“宿主第二基因组”—肠道菌群的影响。在世代传递中,宿主的遗传信息可通过配子稳定遗传。自1918年英国统计学家和遗传学家Fisher[2]提出方差组分剖分思想以来,科研工作者对复杂性状遗传规律的研究,由亲缘个体间表型相似性转变为遗传效应的剖分,再到随机群体全基因组位点贡献解析的过程中,遗传力(heritability)的概念始终贯穿其中。
与宿主遗传取得的一系列突破成果相比,肠道微生物与宿主表型联系的研究进展则缓慢许多。早在1907年,诺贝尔奖获得者Metchnikoff在《The Prolongation of Life: Optimistic Studies》一书中最先明确提出肠道微生物产生的毒素是人体衰老、患病的重要原因,通过调控肠道菌群可以促进健康、延缓衰老[3]。但是由于菌群分离培养和传统的分子生物学方法(如DNA指纹技术)的局限性,他的观点始终未得到主流医学界的关注和认可。近年来,以二代高通量测序技术为主要手段的宏基因组学(metagenomics)及其生物信息分析方法[4]广泛应用于动物肠道菌群的研究,揭示了动物肠道微生物多样性、结构和功能,肠道微生物组的研究成为微生物学乃至整个生物学最具活力的研究领域之一,新发现和新理论如雨后春笋,层出不穷。尤其是近年来各国学者将数量遗传学上的概念、理论和方法引入肠道微生物的研究工作中[5~9],掀起了新一轮的研究热潮。本文系统介绍了近百年来宿主遗传效应估计和肠道菌群效应解析,以及宿主与肠道菌群相互关系(图1)研究进展,并进行简要讨论和展望,以期为该领域的深入研究提供参考。
1 宿主基因组与性状间的关系
1.1 遗传力的定义
遗传力是一个基于数理统计、主要反映宿主数量性状遗传特征的重要参数,习惯上用h表示,该符号最早是1920年美国遗传学家Wright[10]在豚鼠()斑纹的通径分析(path analysis)中,用来表示宿主遗传因素对机体表型的决定程度。一般情况下指狭义遗传力(narrow sense heritability),即加性方差(即育种值方差)占据表型方差的比率,其生物统计学概念等同于加性效应对表型值的回归系数,或加性效应与表型值的相关系数的平方[11]。需要强调的是,世代更迭,只有基因通过配子进行传递,因此遗传力不能真实反映性状的传递能力,仅说明的是群体特定性状的变异情况[12]。
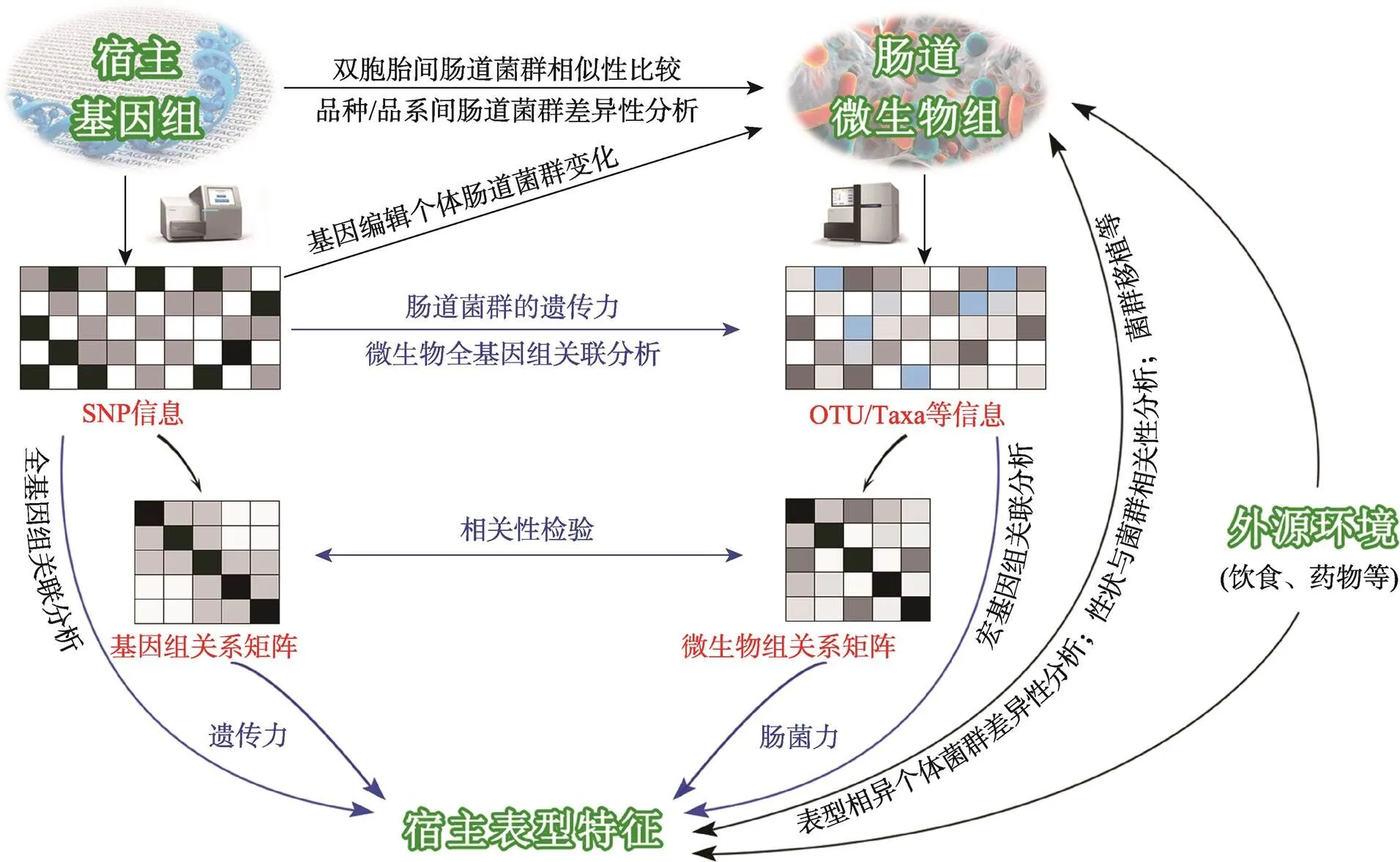
图1 宿主和肠道菌群相互关系及相关研究方法
1.2 遗传力估计方法
由于遗传力反映了亲属间的相似程度,传统数量遗传学通常基于亲缘关系较近的两类个体表型值间的相关系数及亲缘系数来估计遗传力(图2A)[13]。例如子代对任一亲本表型值或中亲值的回归[14]、全同胞或半同胞的相关性[15],以及常用于人类性状的双胞胎间相似性[16]。
上述经典的遗传力估计方法虽然计算较为简单,但只能处理系谱关系简单、均衡的资料。对于系谱关系复杂(如多代和跨代)或者非均衡的群体,通过线性混合模型估计加性遗传方差和环境组分,再利用获得的方差组分计算遗传力则更为有效。1953年美国数量遗传学家Henderson发表了题为《Estimation of variance and covariance components》的论文[17],提出了3种适用于非均衡资料的方差组分估计方法,引起遗传学家和统计学家的广泛关注。为提高方差组分的估计准确性,各国的遗传育种工作者及对此感兴趣的统计学家进行了大量的探索,估计方法也随着新理论、新技术和新工具的不断涌现而更新和发展,如约束最大似然法(restricted maximum likelihood, REML)[18]、贝叶斯估计法(Bayesian method)[19]和Gibbs抽样法[20]等。此后,虽然新方法仍在不断出现,但是REML法和贝叶斯估计法已在全球范围内占据主导地位。
线性模型的理论及其估计方法在现代数量遗传学中占有十分重要的地位,尤其是动物模型,更是畜禽育种、进化遗传学和人类遗传学部分应用中的首选模型[12]。传统上利用线性混合模型估计遗传力都是基于系谱记录完整的群体,因为首先需要利用系谱信息构建分子亲缘相关矩阵(numerator relationship matrix,简称A阵)。由于孟德尔抽样(Mendelian sampling)误差[21],基于系谱推断的个体间亲缘关系准确性有限(图2B),而且系谱又或多或少地存在错误记录;此外,对于系谱信息不完整或缺失的群体以及随机群体,也显得无能为力。理论上,亲缘关系较近的个体,共享的等位基因也更多,由此各国科研工作者积极尝试利用分子标记(如QTL、DNA多态性等)来推断个体间的亲缘关系[22~24]。1996年Ritland[25]首次利用分子标记推断的个体间亲缘关系结合表型相似性估计自然群体的遗传力,此后在植物[26]、鱼[27]、哺乳动物[28]以及人类群体中[29]得到进一步完善和应用。
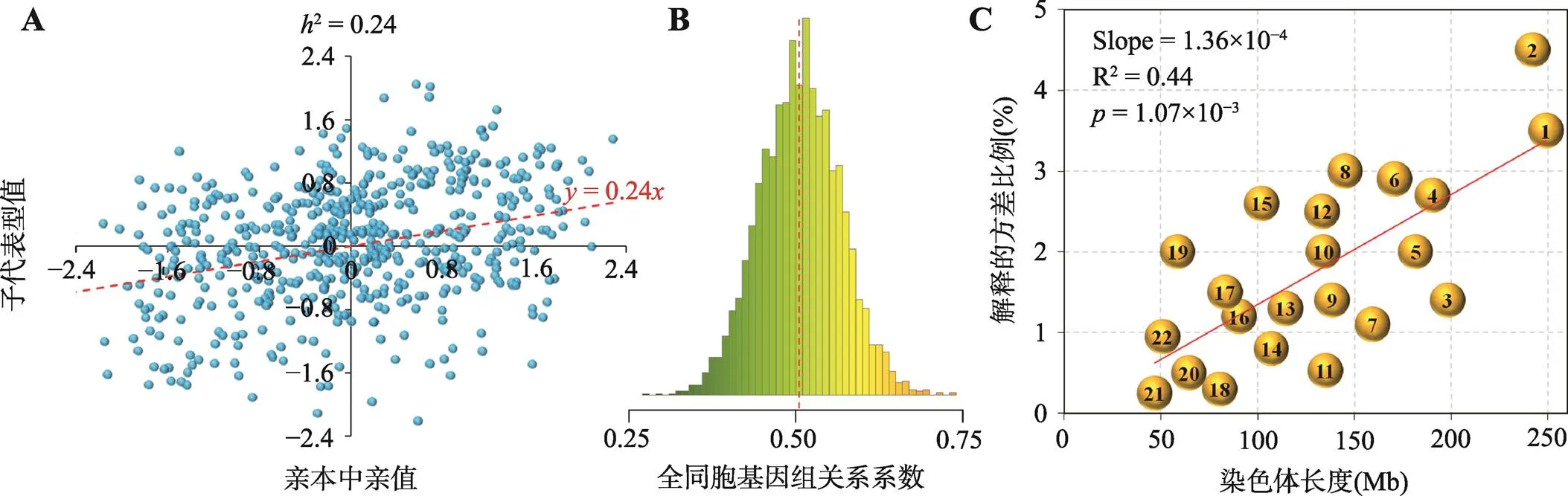
图2 遗传力估计的主要信息来源
A:基于子代表型值对亲本中亲值的回归估计遗传力(根据参考文献[12]修改绘制);B:蛋鸡全同胞个体间的基因组关系系数分布(中间虚线为均值线,由于孟德尔抽样误差,全同胞个体间的亲缘系数可能偏离0.5);C:基于每条染色体上的SNP估计每条染色体所能解释目标性状的遗传方差。

1.3 全基因组关联分析
1996年,美国斯坦福医学院Risch等[42]发现复杂疾病遗传学研究中的关联分析比连锁分析具有更高的检测效力,随即提出全基因关联分析(genome-wide association study, GWAS)的概念:在整个基因组范围内筛选与目标性状相关联的分子标记。基于测序和基因分型技术,2005年Klein等[43]首次报道了一项有关年龄相关性视网膜黄斑变性的GWAS研究。之后,一系列与人类复杂疾病等相关表型的GWAS研究被陆续报道[44~46],并且GWAS在畜禽各种重要经济性状及复杂疾病抗性的遗传研究中也得到了广泛应用[47~49]。GWAS研究为揭示机体复杂性状的遗传奥秘开辟了新的渠道,筛选出大量与目标性状关联的主效和微效基因,并被纳入临床应用[50]和遗传选育[51]。
2 宿主基因组与肠道菌群的关系
动物表型变异除了受宿主自身遗传因素的影响,肠道内栖息的数量庞大而复杂的微生物及其代谢产物的作用亦不能小觑,它们通过肠–肝[52]、肠–脑[53]等调控轴与宿主免疫疾病[54~56]、营养代谢[57~59]和机体行为[60~62]等诸多方面密切联系。肠道菌群究竟是先天遗传因素主导,还是后天环境因素驱动?这一直是肠道微生物研究领域的焦点之一。国内外学者采用各种研究手段和方法,持续追问“nature or nurture?”
双胞胎样本是解析人类基因组塑造肠道菌群的良好模型。早期基于细胞培养[63]和DNA指纹技术[64,65]发现与异卵双胞胎和无亲缘关系个体相比,同卵双胞胎的粪便微生物结构更为相似。近年来,Turnbaugh等[66]和Yatsunenko等[67]通过16S rRNA测序,在稍大规模的双胞胎人群也观察到这一类似结果,但未达到显著水平,这两项研究更多的是强调环境因素在塑造肠道微生物的关键性作用。肠道经历了从少到多、从简单到复杂、从不稳定直至相对稳定的菌群定植过程[67,68],但是并不能确定肠道微生物结构的相似性是由于遗传背景的相似还是由于生活环境一致所造成的。Xie等[69]对250对双胞胎的粪便样品进行宏基因组测序,结果表明人体基因组对肠道内诸多菌群及其潜在功能具有显著贡献,且双胞胎之间,尤其是同卵双胞胎之间的肠道微生物组成、SNP和功能分类高度相似,但是这种相似性又会随着异地居住而逐渐减弱。在小鼠()[70,71]以及鸡()[72~74]、猪()[75]、牛()[76~78]和羊()[79,80]等畜禽上,研究者更多的是比较不同品系和品种在相同饲养条件下、或相同品系/品种在不同饲养条件下的肠道微生物的差异;这些研究表明在相同饲养条件下,肠道菌群具有明显的品种/品系特异性,但饲养环境的差异又掩盖了遗传因素对肠道菌群的塑造作用。
2010年美国内布拉斯加大学Beason等[5]提出将肠道微生物作为宿主的复杂数量性状进行研究。此后,诸多学者将肠道菌群丰度、α和β多样性以及微生物基因功能丰度等视为数量性状(稀有的分类群视为二分类性状),估计它们的遗传力,辨别受宿主遗传因素调控的微生物群落,并进一步通过微生物全基因组关联分析(microbial genome-wide association study, mGWAS)挖掘导致肠道菌群可遗传性的宿主遗传变异。
2.1 肠道菌群的遗传力
将肠道菌群的遗传力理解为肠道微生物组成和相对丰度能够稳定遗传下去的能力是一种误解,更为合适的解释是其反映了宿主遗传因素对肠道微生物的影响程度,或亲缘个体间肠道菌群的相似性[81]。通过对宿主自身基因组和肠道微生物分别采用基因芯片分型和16S rRNA测序的方法,Goodrich等[8]于2014年分析了来自英国的416对双胞胎的粪便样本,结果显示同卵双胞胎相较异卵双胞胎和无亲缘个体具有更加相似的微生物结构,且3组样本间的相似性距离达到显著差异水平;作者进一步通过估计肠道菌群的遗传力,鉴定出33个相对丰度受到宿主遗传因素影响的微生物,尤其是对Christensenellaceae菌科的估计值为0.33,该结果在加拿大(h=0.65)[82]和韩国人群粪便样本(h=0.31)[83]中也得到了验证。考虑到样本数量的影响,2016年Goodrich等[84]将双胞胎数量提高到1126对,对Christensenellaceae的遗传力估计值升至0.42,同时观察到更多的可遗传性微生物(遗传力大于0.2的菌群比例仅由此前的5.3%增加到8.8%),这些微生物的相对丰度在相当长的时间内保持稳定,厚壁菌门(Firmicutes)和放线菌门(Actinobacteria)具有更多的遗传属性,而高丰度的拟杆菌门(Bacteroidetes)呈现较少的可遗传性;除了估计分类群的遗传力外,作者也估计了α和β多样性的遗传力,其中α多样性也具有一定的遗传力。同年,Turpin等[82]在加拿大招募了1561名志愿者,估计了从门至属水平共计249种粪便菌群的遗传力,结果发现94种的细菌类群受宿主遗传背景的影响,其遗传力介于0.25~0.66;Lim等[83]基于655名韩国人体的粪便样本,估计了85种不同分类水平下的菌群遗传力,其中50种菌群具有显著的遗传力,并且双歧杆菌属()所在的放线菌门(Actinobacteria)的遗传力达0.46。
在畜禽上,基于16S rRNA测序数据,肠道菌群遗传力的估计也得到了广泛开展。2013年,Zhao等[85]利用系谱信息估计了弗吉尼亚理工大学体重双选系肉鸡部分粪便菌群的遗传力,证明肉鸡粪便微生物组成受到宿主遗传背景的影响;随后,Meng等[86]还分析了肠道菌群的遗传相关,并表明一些菌群之间存在显著的遗传相关。Wen等[87]估计了肉鸡十二指肠、空肠、回肠和盲肠4个肠段以及粪便微生物的SNP遗传力,发现可遗传微生物主要是厚壁菌门(Firmicutes)和变形菌门(Proteobacteria),但各肠段具有显著SNP遗传力的微生物累积丰度均不到3.5%。在反刍动物上,Sasson等[88]通过50K SNP芯片获取奶牛自身基因组,确定了22个相对丰度与能量捕获的可遗传瘤胃微生物。Difford等[89]基于系谱信息估计了750头荷斯坦奶牛瘤胃细菌和古菌遗传力,结果表明在可操作分类单元(operational taxonomic units, OTUs)上,仅有6%的细菌和12%的古菌具有显著的遗传力;在属水平上,144个细菌属中只有8个具有显著的遗传力估计值(0.17~0.25),此外只有3个古菌属具有遗传力(0.18~0.22)。Li等[90]利用50K SNP芯片对709头肉牛进行基因分型,估计了174个(137个细菌,27个古菌)不同分类水平下的瘤胃微生物SNP遗传力,发现56个细菌和3个古菌受到宿主基因组的调控(h=0.15~0.25),其中22个属于厚壁菌门(Firmicutes),并且细菌和古菌一些衡量α和β多样性的指标也呈现一定的遗传力。Chen等[91]估计了256头二花脸猪盲肠微生物和244头巴马香猪粪便微生物的SNP遗传力,共发现67种盲肠菌群和81种粪便微生物的遗传力估计值高于0.15(两种样本可遗传分类群交集数为31),其中粪便中的菌种遗传力达到0.41,盲肠中的Ruminococcaceae菌科和菌属遗传力达到0.56。Camarinha-Silva等[92]在207头皮特兰母猪上发现49个结肠菌属中8个具有显著的SNP遗传力(0.32~ 0.57),其中菌属的遗传力为0.33。Lu等[93]在大白猪和长白猪杂交后代上的研究表明,粪便微生物α多样性和OTU丰富度在多个时间点具有中等的遗传力。同样,在锯齿动物上也有肠道菌群遗传力的相关报道[94,95]。
2.2 微生物全基因组关联分析
在人类和动物群体中的研究结论虽然有些差异,但均暗示着一部分肠道菌群受到宿主基因组的影响。因此,科研人员进一步通过mGWAS探讨了宿主分子标记与肠道微生物的关联。2015年Blekhman等[9]利用人类微生物组计划(human microbiome project, HMP)数据首次报道了粪便菌群中的双歧杆菌属()与2号染色体上的乳糖酶(lactase,)基因区域之间的关联,乳糖不耐受个体饮用牛奶后,肠道内双歧杆菌的相对丰度升高,以帮助宿主分解乳糖。基因与的关联信号在英国双胞胎群体[84,96]、北美哈特教派信徒[84]、荷兰[97]、德国[98]和HMP[99]队列得到进一步验证,这也是迄今为止mGWAS中报道最一致的信号(表1)。
人类mGWAS研究主要依赖于粪便样品,而畜禽和锯齿动物可以通过活体或者屠宰取样,对粪便以外的其他肠段进行mGWAS研究,并且筛选到一些与肠道微生物丰度和组成显著关联的分子标记(表2)。
除了估计菌群遗传力和进行mWGAS研究外,科研人员也发现基因编辑后的小鼠肠道菌群发生了显著改变[105~108]。这些研究证明了宿主遗传基因型对肠道微生物的作用,但并未回答宿主遗传组分和环境因素对肠道菌群结构的塑造作用究竟有多强这一核心问题。2018年,Rothschild等[96]重新分析了Goodrich等[84]分析的1126对双胞胎数据,结果表明人类基因组组分对肠道微生物组的影响甚微,粪便微生物组的整体可遗传性仅为1.9%。Wang等[98]对1812名德国人群的研究结果显示,遗传因素可解释微生物组β多样性变异的10.43%,而仅年龄、BMI、吸烟与否和性别4个因素的比例可达14.66%;Zhernakova等[109]通过问卷调查的方式收集了荷兰1135名志愿者的饮食、药物使用等多种可能影响肠道微生物的信息,鉴定出几个可影响肠道微生物结构的环境因子,它们可解释18.7%的肠道微生物组β多样性;Rothschild等[96]对1046名以色列人群的研究显示,饮食、药物使用等环境因素可以解释20.03%的肠道微生物组结构变异,而宿主遗传背景与肠道微生物之间没有显著关联。分析宿主亲缘关系与Bray-Curtis相异度相关性的研究结果也表明两者相关性微弱[87,96,103,110]。

表1 人类肠道mGWAS相关研究

表2 畜禽肠道mGWAS相关研究
总之,越来越多的证据表明宿主遗传背景对肠道微生物有影响,但是这种影响对肠道菌群整体塑造作用是有限的(图3)。因此,本文认为肠道微生物组和宿主基因组应该视为不同的实体,结合宿主自身遗传因素,来分析它们对宿主生理性状的影响。
3 肠道菌群与性状间的关系
3.1 肠菌力的概念

图3 宿主遗传信息与肠道菌群的关系
A:宿主自身基因型矩阵和肠道菌群丰度矩阵,部分菌群受到宿主基因型的影响;B:利用A图中的两个矩阵构建的基因组关系矩阵和微生物相似矩阵;C:两两个体间基因组关系系数与Bray-Curtis相异度的散点图,两者相关性不显著。
基于肠菌力的估计值,可以更好的区分各肠段微生物与目标性状的关联程度,进而有针对性的选择特定肠段开展后续研究,尤其是对于可以活体和屠宰取样的畜禽动物,在宏基因组、宏转录组、宏代谢组和宏蛋白质组测序成本依然偏高情况下,先利用16S rRNA测序数据筛选出重要肠段,之后再采用分辨率更高的宏基因组等手段揭示肠道微生物种群及其代谢物、菌群基因及基因产物对机体生理性状的作用机制。肠菌力的估计也为一些传统研究方法的改进提供了依据,如通过肠菌力估计值可以明确禽类盲肠微生物在消化代谢过程中具有的重要作用,因此去盲肠法测定消化率并不能有效反映机体真实的消化率。
3.2 肠菌力的估计方法
3.2.1 方差组分法
估计肠菌力最早的研究可追溯至2013年,澳大利亚的研究者[7]借鉴宿主SNP遗传力的估计方法,利用宏基因组测序得到的重叠群计数(contig count)数据构建微生物组关系矩阵(metagenomic relationship matrix,也有研究使用microbial relationship matrix和bacterial kinship matrix,后文统称M阵,表4),接着采用线性混合模型评估奶牛瘤胃微生物组对甲烷排放以及人类粪便微生物组对BMI和IBD的贡献大小。此后,Difford等[89]采用该方法估计了750头荷斯坦奶牛甲烷排放的遗传力和肠菌力(分别基于系谱信息和16S rRNA测序),结果表明宿主遗传和瘤胃微生物对奶牛甲烷排放的效应值分别为0.21和0.13,且相互独立。2017年,德国霍恩海姆大学Camarinha-Silva等[92]利用16S rRNA测序和Illumina 60K芯片分别分析了207头皮特兰母猪结肠微生物组成和宿主基因型,基于线性混合模型分别估算了日增重、采食量和饲料转化率的微生物效应和遗传效应,结果显示3个性状的遗传力分别为0.35、0.20和0.23,而肠菌力亦分别可达0.41、0.33和0.33。与澳大利亚学者不同的是,后两项研究使用的是OTU相对丰度构建M阵。
2018年,以色列魏兹曼科学研究所Rothschild等[96]收集了1046名以色列人的身高、BMI、胸围、臀围、各血液生化指标等12个复杂性状,对粪便微生物采用了宏基因组和16S两种测序手段。基于宏基因组测序得到的微生物基因检出与否(0/1)构建M阵,将宿主性别、年龄、饮食以及前5个遗传主成分作为协变量,使用GCTA估计微生物组对宿主各表型的可释方差(microbiome-association index, b2)。此外,该研究利用基因芯片对宿主基因组进行分型,提取已公布的与各目标性状相关的SNP位点,计算目标性状的多基因风险评分(polygenic risk score, PRS),将PRS或者SNP基因型作为协变量加入线性混合模型中以校正宿主遗传效应的影响,结果显示肠道微生物组对12个性状中的8个具有显著的影响,并且对于某些性状,微生物效应大小不亚于遗传效应。该研究还发现估计肠菌力所需样本数远小于遗传力的样本需求量。在该研究中,除了利用微生物基因检出与否构建M阵外,作者也尝试利用基因丰度构建M阵,以及基于两种测序方式聚类得到的各分类水平下的菌群丰度或检出与否构建M阵。遗憾的是作者并未使用OTU数据构建M阵,也未对基于不同数据所构建的M阵效果进行讨论,但从该研究公布的数据看,由于分类单元信息量过少,估计的效果并不理想,而使用基因检出与否和相对丰度的估计效果差异相对较小。

表3 人类和动物上肠菌力估计的相关研究
值得注意的是,上述几项有关肠菌力估计的研究仅基于一个肠段内容物。然而,大量的研究表明,由于各肠段间的氧气含量[114]、pH值[115]和营养物质可用性[116]存在空间异质性,各个肠段的微生物群落组成、结构和功能亦具有较大的差异[87,117]。此外,宿主自身基因组对目标性状都具有较大的影响,而在估计各性状肠菌力时,科研人员并没有校正宿主遗传效应,或者校正依据的是公共数据库中与目标性状相关联的位点,而不是待估群体自身的关联位点。
由此,本课题组以205只黄羽肉鸡为研究对象,对宿主进行全基因组重测序(whole genomic resequencing, WGS),通过全基因组关联分析筛选与腹脂量和腹脂率关联的位点;并对宿主十二指肠、空肠、回肠、盲肠及粪便微生物进行16S rRNA测序,利用各段OTU相对丰度数据分别构建M阵,通过线性混合模型依次估计各肠段和粪便的微生物对腹脂沉积的效应[87]。考虑到宿主自身遗传基础在腹脂沉积的过程中发挥着较大作用,在估计肠菌力时,提取与腹脂沉积关联的位点基因型,通过降维选择前两个主成分以及前5个遗传主成分作为协变量以校正遗传因素干扰。结果表明,腹脂量在十二指肠、空肠、回肠、盲肠、粪便的肠菌力依次为:0.24、0.06、0.03、0.21和0.02,腹脂率在5个部分的肠菌力分别为:0.24、0.10、0、0.20和0.03,这意味着不同肠段的微生物组对腹脂沉积的贡献是有差异的,该结果为研究者选择肠段开展后续研究提供了重要参考。
综上所述,方差组分法主要借鉴了SNP遗传力估计的方法,通过线性混合模型求解。方差组分法的核心在于M阵的构建,虽然现有的几篇文献在M阵构建的基本形式一致(公式(1)),但是在选用于构建M阵的数据类型并不完全一致(图4),本文对其进行了系统比较,以期为将来肠菌力估计提供参考。

式中,X为×的矩阵,XT为X的转置矩阵,为个体数,为检测到的OTU(或conting/gene)总数。X任一元素x为个体的第个OTU (或conting/ gene)的标准化的相对丰度(或标准化的contig计数、gene检出与否)。M阵中个体和的微生物关系系数m计算公式如表4所示。
目前,通过宏基因组和扩增子两种测序方法得到的肠道微生物数据都已用于估计肠菌力,前者分辨率高、获取的信息量大,不仅能提供菌群群落层面的信息,也能揭示微生物组功能基因信息;而后者成本低廉,分析快速,且能有效避免宿主基因组信息的污染,但这种测序方式需要经过PCR扩增,研究范围受限于可扩增部分序列的物种组成,因此会遗漏一些菌群信息。由于缺少实际对比研究,基于不同测序方法的肠菌力评估效果还有待后续进一步探讨。
3.2.2 回归法
除方差组分法外,2015年荷兰格罗宁根大学Fu等[118]为估计肠道微生物组对人体血脂含量和BMI的贡献大小,对荷兰LifeLines-DEEP项目中893名参与者的粪便微生物进行16S rRNA测序,提出通过二分类模型(binary model,估计OTU检出与否的评分)和数量模型(quantitative model,估计OTU相对丰度的评分)分别估计每个OTU两种不同的属性的风险评分,再依据加性模型(additive model)求解每个个体所有OTU累加的微生物风险评分,并将其与个体表型值相关系数的平方(the squared correlation coefficient, R2)作为肠道微生物组对宿主表型的可释方差。虽然该方法主要借鉴了流行病学研究中的遗传风险预测模型[119,120],但其与数量遗传学中狭义遗传力的估计(加性效应与表型值相关系数的平方[11])有相似之处。该方法不涉及M阵的构建,先估计每种OTU效应的大小,再计算个体OTU累加的总效应,最后以OTU总效应与表型值相关系数的平方作为肠菌力。该方法为评估每类OTU效应,以及每个个体肠道微生物效应值提供了重要参考,但尚无实际数据分析回归法与方差组分法的估计效果,其实际应用还需要更深入探索和评估。

表4 微生物组关系矩阵构建方法比较

图4 基于高通量测序微生物组关系矩阵构建流程
3.3 宏基因组关联分析
为了能够高分辨率的研究肠道微生物组与宿主表型的关联,Qin等[121]借鉴GWAS研究,于2012年提出了宏基因组关联分析(metagenome-wide association study, MWAS)的概念和方法。该研究对345个中国参与者的粪便微生物进行了两阶段MWAS,共鉴定出约6万个与Ⅱ型糖尿病相关的分子标记,并从菌种、功能以及微生态群落详尽展示了肠道微生物与宿主表型的关联特征。
通过全集因组鸟枪法测序,将质控后的高质量序列拼接成更长的重叠群,通过对重叠群进行基因预测,去除样品间高度相似的基因序列,得到高质量的非冗余参考基因集;基于来源于同一菌株基因组的重叠群或基因在不同样品中应具有一致的丰度变化模式,对宏基因组数据进行聚类,将物种分辨率提高到菌株水平[122],这些聚类方法包括建立宏基因组连锁群(metagenomic linkage group, MLG)[121]、宏基因组基因簇(metagenomic clusters, MGC)[123]和宏基因组物种基因群组(metagenomic species, MGS)[124]。特别是2014年Nielsen等[124]提出的MGS方法,可以在不依赖参考基因集的情况下,将微生物基因组鸟枪法测序数据转化成微生物物种信息。
MWAS不仅能鉴定相对丰度发生变化的细菌种类,基于KEGG、COG和EggNOG等数据库对基因序列进行功能聚类,还能判断菌群功能的增强与减弱。原则上MWAS可用于研究微生物组与任何性状之间的关联,但迄今为止还主要集中在II型糖尿病[121]、肥胖[125]、结直肠癌[126]以及类风湿性关节炎[127]等病例对照设计研究上。随着微生物领组领域的发展,期待未来MWAS在复杂的数量性状上能得到广泛的应用。
4 结语与展望
历经百年的发展和累积,遗传力的估计方法不断改进、丰富和完善。将遗传效应的分析模型和统计方法引入肠道微生物的研究中,为肠道微生物、宿主基因组和宿主表型特征的关系研究提供了全新的思路。尽管在该方向上取得了显著的进展,但尚处于早期阶段,仍然具有挑战,主要表现在5个方面:(1)肠菌力的准确评估,以及宿主基因组与肠道菌群互作机制的解析,都需要足够的样本量,与人类上的研究相比,畜禽上的相关研究所使用的样本数量相对较少,在测序成本以“超摩尔定律”的速度不断下降的同时,应适当扩大分析的样本含量,以增强结果的可靠性;(2)不管是宏基因组测序还是扩增子测序,通过序列相似度进行聚类,都存在归类不准确的现象,进而导致定量的偏差;(3)肠道微生物组是一个复杂的多维结构,基于两种测序数据计算肠道菌群的遗传力、鉴定与菌群显著关联的分子标记和评估肠菌力时,是否需要考虑微生物之间的互作关系,以及对数据进行质控过滤的条件还有待进一步探讨;(4)目前对OTU、重叠群或基因等数据的属性尚无统一标准,应该将其视为二分类属性(0/1),还是连续变异,亦或是将两者同时纳入考虑,它们的估计效果又是否有差异亦有待深入论证; (5)尤为重要的一点是,肠道微生物对外界环境因素高度敏感,外源环境因素可能会掩盖宿主遗传变异对肠道菌群的影响,并且也会造成肠菌力评估和MWAS结果偏差。
随着测序技术的快速发展,微生物组分析技术、工具和高质量的参考数据库将升级换代,随之而来的肠道微生物分析所需要的数据量、深度和精度将不再成为瓶颈。受回归法估计肠菌力的启示,或许未来人们可以像估计SNP效应值一样,估计OTU、重叠群或微生物基因的效应值,以确定哪一类菌群的哪一簇基因与目标性状关系更为紧密;同时将微生物效应值作为协变量加入到模型中以提高遗传评估的准确性,如在人类疾病预测上,综合考虑PRS和微生物效应可更为准确的预测疾病易感风险。但如何在mGWAS、肠菌力评估和MWAS过程中调整环境因素的影响是一大挑战,我们期待结合数量遗传学的最新理论和方法,肠菌力的估计会被进一步完善,并将取得更多、更有影响的创新和突破。
[1] Rosenberg E, Zilber-Rosenberg I. The hologenome concept of evolution after 10 years., 2018, 6(1): 78.
[2] Fisher RA. The correlation between relatives on the supposition of Mendelian inheritance., 1919, 52(02): 399–433.
[3] Podolsky SH. Metchnikoff and the microbiome., 2012, 380(9856): 1810–1811.
[4] Liu YX, Qin Y, Guo XX, Bai Y. Methods and applications for microbiome data analysis., 2019, 41 (9): 845–862.刘永鑫, 秦媛, 郭晓璇, 白洋. 微生物组数据分析方法与应用. 遗传, 2019, 41 (9): 845–862.
[5] Benson AK, Kelly SA, Legge R, Ma F, Low S J, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, Kachman SD, Moriyama EN, Walter J, Peterson DA, Pomp D. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors., 2010, 107(44): 18933–18938.
[6] Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto J, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J. A metagenome-wide association study of gut microbiota in type 2 diabetes., 2012, 490(7418): 55–60.
[7] Ross EM, Moate PJ, Marett LC, Cocks BG, Hayes BJ. Metagenomic predictions: from microbiome to complex health and environmental phenotypes in humans and cattle., 2013, 8(9): e73056.
[8] Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, Spector TD, Clark AG, Ley RE. Human genetics shape the gut microbiome., 2014, 159(4): 789–799.
[9] Blekhman R, Goodrich JK, Huang K, Sun Q, Bukowski R, Bell JT, Spector TD, Keinan A, Ley RE, Gevers D, Clark AG. Host genetic variation impacts microbiome composition across human body sites., 2015, 16(1): 191.
[10] Wright S. The relative importance of heredity and environment in determining the piebald pattern of guinea-pigs., 1920, 6(6): 320– 332.
[11] Falconer DS, Mackay TFC. Introduction to Quantitative Genetics, Fourth Edition: Longman Press, 1996, 160– 181.
[12] Visscher PM, Hill WG, Wray NR. Heritability in the genomics era--concepts and misconceptions., 2008, 9(4): 255–266.
[13] Lstibůrek M, Bittner V, Hodge GR, Picek J, Mackay TFC. Estimating realized heritability in panmictic populations., 2018, 208(1): 89–95.
[14] Robertson FW. Studies in quantitative inheritance XI. Genetic and environmental correlation between body size and egg production in., 1957, 55(3): 428.
[15] Fredeen HT, Jonsson P. Genic variance and covariance in Danish Landrace swine as evaluated under a system of individual feeding of progeny test group., 1957, 70(4): 348–363.
[16] Boomsma D, Busjahn A, Peltonen L. Classical twin studies and beyond., 2002, 3(11): 872–882.
[17] Henderson CR. Estimation of variance and covariance components., 1953,(9): 226–252.
[18] Patterson HD, Thompson R. Recovery of inter-block information when block sizes are unequal., 1971, 58(3): 545–554.
[19] Gianola D, Fernando RL. Bayesian methods in animal breeding theory., 1986, 63(1): 217–244.
[20] Geman S, Geman D. Stochastic relaxation, gibbs distributions, and the bayesian restoration of images., 1984, 6(6): 721–741.
[21] Hill WG. Applications of population genetics to animal breeding, from Wright, Fisher and Lush to genomic prediction., 2014, 196(1): 1–16.
[22] Thompson EA. The estimation of pairwise relationships., 1975, 39(2): 173–188.
[23] Lynch M. Estimation of relatedness by DNA fingerprinting., 1988, 5(5): 584–599.
[24] Queller DC, Goodnight KF. Estimating relatedness using genetic markers., 1989, 43(2): 258–275.
[25] Ritland K. Marker-based method for inferences about quantitative inheritance in natural populations., 1996, 50(3): 1062–1073.
[26] Ritland K, Ritland C. Inferences about quantitative inheritance based on natural population structure in the yellow monkeyflower,., 1996, 50(3): 1074–1082.
[27] Mousseau TA, Ritland K, Heath DD. A novel method for estimating heritability using molecular markers., 1998, 80(2): 218–224.
[28] Thomas SC, Coltman DW, Pemberton JM. The use of marker-based relationship information to estimate the heritability of body weight in a natural population: a cautionary tale., 2002, 15(1): 92–99.
[29] Visscher PM, Medland SE, Ferreira MAR, Morley KI, Zhu G, Cornes BK, Montgomery GW, Martin NG. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings., 2006, 2(3): e41.
[30] Li H, Glusman G, Hu H, Shankaracharya, Caballero J, Hubley R, Witherspoon D, Guthery SL, Mauldin DE, Jorde LB, Hood L, Roach JC, Huff CD. Relationship estimation from whole-genome sequence data., 2014, 10(1): e1004144.
[31] Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, Goddard ME, Visscher PM. Common SNPs explain a large proportion of the heritability for human height., 2010, 42(7): 565–569.
[32] Yang J, Zeng J, Goddard ME, Wray NR, Visscher PM. Concepts, estimation and interpretation of SNP-based heritability., 2017, 49(9): 1304–1310.
[33] Benjamin DJ, Cesarini D, van der Loos MJHM, Dawes CT, Koellinger PD, Magnusson PKE, Chabris CF, Conley D, Laibson D, Johannesson M, Visscher PM. The genetic architecture of economic and political preferences., 2012, 109(21): 8026–8031.
[34] Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis., 2011, 88(1): 76–82.
[35] Yang J, Manolio TA, Pasquale LR, Boerwinkle E, Caporaso N, Cunningham JM, de Andrade M, Feenstra B, Feingold E, Hayes MG, Hill WG, Landi MT, Alonso A, Lettre G, Lin P, Ling H, Lowe W, Mathias RA, Melbye M, Pugh E, Cornelis MC, Weir BS, Goddard ME, Visscher PM. Genome partitioning of genetic variation for complex traits using common SNPs., 2011, 43(6): 519–525.
[36] Davies G, Tenesa A, Payton A, Yang J, Harris SE, Liewald D, Ke XY, Le Hellard S, Christoforou A, Luciano M, McGhee K, Lopez L, Gow AJ, Corley J, Redmond P, Fox HC, Haggarty P, Whalley LJ, McNeill G, Goddard ME, Espeseth T, Lundervold AJ, Reinvang I, Pickles A, Steen VM, Ollier W, Porteous DJ, Horan M, Starr JM, Pendleton N, Visscher PM, Deary IJ. Genome-wide association studies establish that human intelligence is highly heritable and polygenic., 2011, 16(10): 996–1005.
[37] Vinkhuyzen AAE, Pedersen NL, Yang J, Lee SH, Magnusson PKE, Iacono WG, McGue M, Madden PAF, Heath AC, Luciano M, Payton A, Horan M, Ollier W, Pendleton N, Deary IJ, Montgomery GW, Martin NG, Visscher PM, Wray NR. Common SNPs explain some of the variation in the personality dimensions of neuroticism and extraversion., 2012, 2(4): e102.
[38] Loh P, Bhatia G, Gusev A, Finucane HK, Bulik- Sullivan BK, Pollack SJ, de Candia TR, Lee SH, Wray NR, Kendler KS, O'Donovan MC, Neale BM, Patterson N, Price AL. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis., 2015, 47(12): 1385–1392.
[39] Yuan J, Sun C, Dou T, Yi G, Qu L, Qu L, Wang K, Yang N. Identification of promising mutants associated with egg production traits revealed by genome-wide association study., 2015, 10(10): e0140615.
[40] Mathew B, Léon J, Sillanpää M J. A novel linkage- disequilibrium corrected genomic relationship matrix for SNP-heritability estimation and genomic prediction., 2018, 120(4): 356–368.
[41] McCoy AM, Norton EM, Kemper AM, Beeson SK, Mickelson JR, McCue ME. SNP-based heritability and genetic architecture of tarsal osteochondrosis in North American Standardbred horses., 2019, 50(1): 78–81.
[42] Risch N, Merikangas K. The future of genetic studies of complex human diseases., 1996, 273(5281): 1516–1517.
[43] Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration., 2005, 308(5720): 385– 389.
[44] Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JRB, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennett AJ, Melzer D, Ferrucci L, Loos RJF, Barroso I, Wareham NJ, Karpe F, Owen KR, Cardon LR, Walker M, Hitman GA, Palmer CNA, Doney ASF, Morris AD, Smith GD, Hattersley AT, McCarthy MI. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity., 2007, 316(5826): 889–894.
[45] Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, Hughes TE, Groop L, Altshuler D, Almgren P, Florez JC, Meyer J, Ardlie K, Bengtsson BK, Isomaa B, Lettre G, Lindblad U, Lyon HN, Melander O, Newton-Cheh C, Nilsson P, Orho-Melander M, Rastam L, Speliotes EK, Taskinen M R, Tuomi T, Guiducci C, Berglund A, Carlson J, Gianniny L, Hackett Råstam L, Speliotes EK, Taskinen MR, Tuomi T, Guiducci C, Berglund A, Carlson J, Gianniny L, Hackett R, Hall L, Holmkvist J, Laurila E, Sjögren M, Sterner M, Surti A, Svensson M, Svensson M, Tewhey R, Blumenstiel B, Parkin M, Defelice M, Barry R, Brodeur W, Camarata J, Chia N, Fava M, Gibbons J, Handsaker B, Healy C, Nguyen K, Gates C, Sougnez C, Gage D, Nizzari M, Gabriel SB, Chirn GW, Ma Q, Parikh H, Richardson D, Ricke D, Purcell S. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels., 2007, 316(5829): 1331–1336.
[46] Long T, Hicks M, Yu HC, Biggs WH, Kirkness EF, Menni C, Zierer J, Small KS, Mangino M, Messier H, Brewerton S, Turpaz Y, Perkins BA, Evans AM, Miller LAD, Guo LN, Caskey CT, Schork NJ, Garner C, Spector TD, Venter JC, Telenti A. Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites., 2017, 49(4): 568–578.
[47] Daetwyler HD, Capitan A, Pausch H, Stothard P, van Binsbergen R, Brøndum RF, Liao X, Djari A, Rodriguez SC, Grohs C, Esquerré D, Bouchez O, Rossignol MN, Klopp C, Rocha D, Fritz S, Eggen A, Bowman PJ, Coote D, Chamberlain AJ, Anderson C, VanTassell CP, Hulsegge I, Goddard ME, Guldbrandtsen B, Lund MS, Veerkamp RF, Boichard DA, Fries R, Hayes BJ. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle., 2014, 46(8): 858–865.
[48] Zhang T, Wang WH, Zhang GX, Wang JY, Xue Q, Gu YP. A genome-wide association study on body weight traits of Jinghai yellow chicken,, 2015, 37(8): 811–820.张涛, 王文浩, 张跟喜, 王金玉, 薛倩, 顾玉萍. 京海黄鸡体重性状全基因组关联分析. 遗传, 2015, 37(8): 811–820.
[49] Sun CJ, Qu L, Yi GQ, Yuan JW, Duan ZY, Shen MM, Qu LJ, Xu GY, Wang KH, Yang N. Genome-wide association study revealed a promising region and candidate genes for eggshell quality in an F2 resource population., 2015, 16(1): 565.
[50] Manolio TA. Bringing genome-wide association findings into clinical use., 2013, 14(8): 549–558.
[51] Lopes MS, Bovenhuis H, van Son M, Nordbø Ø, Grindflek EH, Knol EF, Bastiaansen JWM. Using markers with large effect in genetic and genomic predictions., 2017, 95(1): 59–71.
[52] Tripathi A, Debelius J, Brenner D A, Karin M, Loomba R, Schnabl B, Knight R. The gut–liver axis and the intersection with the microbiome., 2018, 15(7): 397–411.
[53] Cryan JF, O'Riordan KJ, Cowan C, Sandhu KV, Bastiaanssen T, Böhme M, Codagnone MG, Cussotto S, Fülling C, Golubeva AV, Guzzetta KE, Jaggar M, Long-Smith CM, Lyte JM, Martin JA, Molinero-Perez A, Moloney G, Morelli E, Morillas E, O'Connor R, Cruz-Pereira J, Peterson VL, Rea K, Ritz NL, Sherwin E, Spichak S, Teichman E, van de Wouw M, Ventura- Silva P, Wallace-Fitzsimons S, Hyland N, Clarke G, Dinan TG. The microbiota-gut-brain axis., 2019, 99(4): 1877–2013.
[54] Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view., 2012, 148(6): 1258–1270.
[55] Gensollen T, Iyer SS, Kasper DL, Blumberg RS. How colonization by microbiota in early life shapes the immune system., 2016, 352(6285): 539–544.
[56] Sanna S, van Zuydam NR, Mahajan A, Kurilshikov A, Vich Vila A, Võsa U, Mujagic Z, Masclee AAM, Jonkers DMAE, Oosting M, Joosten LAB, Netea MG, Franke L, Zhernakova A, Fu J, Wijmenga C, McCarthy MI. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases., 2019, 51(4): 600–605.
[57] Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest., 2006, 444(7122): 1027–1131.
[58] Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions., 2012, 336(6086): 1262–1267.
[59] Martinez-Guryn K, Hubert N, Frazier K, Urlass S, Musch MW, Ojeda P, Pierre JF, Miyoshi J, Sontag TJ, Cham CM, Reardon CA, Leone V, Chang EB. Small intestine microbiota regulate host digestive and absorptive adaptive responses to dietary lipids., 2018, 23(4): 458–469.
[60] Schretter CE, Vielmetter J, Bartos I, Marka Z, Marka S, Argade S, Mazmanian SK. A gut microbial factor modulates locomotor behaviour in., 2018, 563(7731): 402–406.
[61] Rhoades JL, Nelson JC, Nwabudike I, Yu SK, McLachlan IG, Madan GK, Abebe E, Powers JR, Colón-Ramos DA, Flavell SW. ASICs mediate food responses in an enteric serotonergic neuron that controls foraging behaviors., 2019, 176(1–2): 85–97.
[62] Sharon G, Cruz NJ, Kang DW, Gandal MJ, Wang B, Kim YM, Zink EM, Casey CP, Taylor BC, Lane CJ, Bramer LM, Isern NG, Hoyt DW, Noecker C, Sweredoski MJ, Moradian A, Borenstein E, Jansson JK, Knight R, Metz TO, Lois C, Geschwind DH, Krajmalnik- Brown R, Mazmanian SK. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice., 2019, 177(6): 1600–1618.
[63] Van de Merwe JP, Stegeman JH, Hazenberg MP. The resident faecal flora is determined by genetic characterristics of the host. Implications for Crohn's disease?, 1983, 49(2): 119–124.
[64] Zoetendal EG, Akkermans ADL, Akkermans-van Vliet WM, de Visser JAGM, de Vos WM . The host genotype affects the bacterial community in the human gastrointestinal tract., 2001, 13(3): 129–134.
[65] Stewart JA, Chadwick VS, Murray A. Investigations into the influence of host genetics on the predominant eubacteria in the faecal microflora of children., 2005, 54(12): 1239–1242.
[66] Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. A core gut microbiome in obese and lean twins., 2009, 457(7228): 480–484.
[67] Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon JI. Human gut microbiome viewed across age and geography., 2012, 486(7402): 222–227.
[68] Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J, Knights D, Gajer P, Ravel J, Fierer N, Gordon JI, Knight R. Moving pictures of the human microbiome., 2011, 12(5): R50.
[69] Xie H, Guo R, Zhong H, Feng Q, Lan Z, Qin B, Ward KJ, Jackson MA, Xia Y, Chen X, Chen B, Xia H, Xu C, Li F, Xu X, Al-Aama JY, Yang H, Wang J, Kristiansen K, Wang J, Steves CJ, Bell JT, Li J, Spector TD, Jia H. Shotgun metagenomics of 250 adult twins reveals genetic and environmental impacts on the gut microbiome., 2016, 3(6): 572–584.
[70] Friswell MK, Gika H, Stratford IJ, Theodoridis G, Telfer B, Wilson ID, McBain AJ. Site and strain-specific variation in gut microbiota profiles and metabolism in experimental mice., 2010, 5(1): e8584.
[71] Xiao L, Feng Q, Liang S, Sonne SB, Xia Z, Qiu X, Li X, Long H, Zhang J, Zhang D, Liu C, Fang Z, Chou J, Glanville J, Hao Q, Kotowska D, Colding C, Licht TR, Wu D, Yu J, Sung JJY, Liang Q, Li J, Jia H, Lan Z, Tremaroli V, Dworzynski P, Nielsen HB, Bäckhed F, Doré J, Le Chatelier E, Ehrlich SD, Lin JC, Arumugam M, Wang J, Madsen L, Kristiansen K. A catalog of the mouse gut metagenome., 2015, 33(10): 1103–1108.
[72] Ding J, Zhao L, Wang L, Zhao W, Zhai Z, Leng L, Wang Y, He C, Zhang Y, Zhang H, Li H, Meng H. Divergent selection-induced obesity alters the composition and functional pathways of chicken gut microbiota., 2016, 48(1): 93.
[73] Yang L, Liu S, Ding J, Dai R, He C, Xu K, Honaker CF, Zhang Y, Siegel P, Meng H. Gut microbiota co-microevolution with selection for host humoral immunity., 2017, 8: 1243.
[74] Pandit RJ, Hinsu AT, Patel NV, Koringa PG, Jakhesara SJ, Thakkar JR, Shah TM, Limon G, Psifidi A, Guitian J, Hume DA, Tomley FM, Rank DN, Raman M, Tirumurugaan KG, Blake DP, Joshi CG. Microbial diversity and community composition of caecal microbiota in commercial and indigenous Indian chickens determined using 16s rDNA amplicon sequencing., 2018, 6(1): 115.
[75] Xiao Y, Kong F, Xiang Y, Zhou W, Wang J, Yang H, Zhang G, Zhao J. Comparative biogeography of the gut microbiome between Jinhua and Landrace pigs., 2018, 8(1): 5985.
[76] King EE, Smith RP, St-Pierre B, Wright AG. Differences in the rumen methanogen populations of lactating Jersey and Holstein dairy cows under the same diet regimen., 2011, 77(16): 5682–5687.
[77] Gonzalez-Recio O, Zubiria I, García-Rodríguez A, Hurtado A, Atxaerandio R. Short communication: Signs of host genetic regulation in the microbiome composition in 2 dairy breeds: Holstein and Brown Swiss., 2018, 101(3): 2285–2292.
[78] Li FY, Hitch TCA, Chen YH, Creevey CJ, Guan LL. Comparative metagenomic and metatranscriptomic analyses reveal the breed effect on the rumen microbiome and its associations with feed efficiency in beef cattle., 2019, 7(1): 6.
[79] Shi PJ, Meng K, Zhou ZG, Wang YR, Diao QY, Yao B. The host species affects the microbial community in the goat rumen., 2008, 46(1): 132–135.
[80] Douglas JL, Worgan HJ, Easton GL, Poret L, Wolf BT, Edwards A, Davies E, Ross D, McEwan NR. Microbial diversity in the digestive tract of two different breeds of sheep., 2016, 120(5): 1382–1389.
[81] Goodrich JK, Davenport ER, Waters JL, Clark AG, Ley RE. Cross-species comparisons of host genetic associations with the microbiome., 2016, 352(6285): 532– 535.
[82] Turpin W, Espin-Garcia O, Xu W, Silverberg MS, Kevans D, Smith MI, Guttman DS, Griffiths A, Panaccione R, Otley A, Xu L, Shestopaloff K, Moreno-Hagelsieb G, Paterson AD, Croitoru K. Association of host genome with intestinal microbial composition in a large healthy cohort., 2016, 48(11): 1413–1417.
[83] Lim MY, You HJ, Yoon HS, Kwon B, Lee JY, Lee S, Song Y, Lee K, Sung J, Ko G. The effect of heritability and host genetics on the gut microbiota and metabolic syndrome., 2017, 66(6): 1031–1038.
[84] Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, Ober C, Spector TD, Bell JT, Clark AG, Ley RE. Genetic determinants of the gut microbiome in UK twins., 2016, 19(5): 731–743.
[85] Zhao L, Wang G, Siegel P, He C, Wang H, Zhao W, Zhai Z, Tian F, Zhao J, Zhang H, Sun Z, Chen W, Zhang Y, Meng H. Quantitative genetic background of the host influences gut microbiomes in chickens., 2013, 3(1): 1163.
[86] Meng H, Zhang Y, Zhao L, Zhao WJ, He C, Honaker C F, Zhai ZX, Sun ZK, Siegel PB. Body weight selection affects quantitative genetic correlated responses in gut microbiota., 2014, 9(3): e89862.
[87] Wen CL, Yan W, Sun CJ, Ji CJ, Zhou QQ, Zhang DX, Zheng JX, Yang N. The gut microbiota is largely independent of host genetics in regulating fat deposition in chickens., 2019, 13(6): 1422–1436.
[88] Sasson G, Ben-Shabat SK, Seroussi E, Doron-Faigenboim A, Shterzer N, Yaacoby S, Berg MM, White BA, Halperin E, Mizrahi I. Heritable bovine rumen bacteria are phylogenetically related and correlated with the cow's capacity To harvest energy from its feed., 2017, 8(4): e00703–17.
[89] Difford GF, Plichta DR, Løvendahl P, Lassen J, Noel SJ, Højberg O, Wright AG, Zhu Z, Kristensen L, Nielsen HB, Guldbrandtsen B, Sahana G. Host genetics and the rumen microbiome jointly associate with methane emissions in dairy cows., 2018, 14(10): e1007580.
[90] Li FY, Li CX, Chen YY, Liu JH, Zhang CY, Irving B, Fitzsimmons C, Plastow G, Guan LL. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle., 2019, 7(1): 92.
[91] Chen CY, Huang XC, Fang SM, Yang H, He MZ, Zhao YZ, Huang LS. Contribution of host genetics to the variation of microbial composition of cecum lumen and feces in pigs., 2018, 9: 2626.
[92] Camarinha-Silva A, Maushammer M, Wellmann R, Vital M, Preuss S, Bennewitz J. Host genome influence on gut microbial composition and microbial prediction of complex traits in pigs., 2017, 206(3): 1637–1644.
[93] Lu D, Tiezzi F, Schillebeeckx C, McNulty NP, Schwab C, Shull C, Maltecca C. Host contributes to longitudinal diversity of fecal microbiota in swine selected for lean growth., 2018, 6(1): 4.
[94] Org E, Parks BW, Joo JW, Emert B, Schwartzman W, Kang EY, Mehrabian M, Pan C, Knight R, Gunsalus R, Drake TA, Eskin E, Lusis AJ. Genetic and environmental control of host-gut microbiota interactions., 2015, 25(10): 1558–1569.
[95] O’Connor A, Quizon PM, Albright JE, Lin FT, Bennett BJ. Responsiveness of cardiometabolic-related microbiota to diet is influenced by host genetics., 2014, 25(11–12): 583–599.
[96] Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, Costea PI, Godneva A, Kalka IN, Bar N, Shilo S, Lador D, Vila AV, Zmora N, Pevsner- Fischer M, Israeli D, Kosower N, Malka G, Wolf BC, Avnit-Sagi T, Lotan-Pompan M, Weinberger A, Halpern Z, Carmi S, Fu J, Wijmenga C, Zhernakova A, Elinav E, Segal E. Environment dominates over host genetics in shaping human gut microbiota., 2018, 555(7695): 210–215.
[97] Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, Vila AV, Deelen P, Vatanen T, Schirmer M, Smeekens SP, Zhernakova DV, Jankipersadsing SA, Jaeger M, Oosting M, Cenit MC, Masclee AAM, Swertz MA, Li Y, Kumar V, Joosten L, Harmsen H, Weersma RK, Franke L, Hofker MH, Xavier RJ, Jonkers D, Netea MG, Wijmenga C, Fu J, Zhernakova A. The effect of host genetics on the gut microbiome., 2016, 48(11): 1407–1412.
[98] Wang J, Thingholm LB, Skiecevičienė J, Rausch P, Kummen M, Hov JR, Degenhardt F, Heinsen FA, Rühlemann MC, Szymczak S, Holm K, Esko T, Sun J, Pricop-Jeckstadt M, Al-Dury S, Bohov P, Bethune J, Sommer F, Ellinghaus D, Berge RK, Hübenthal M, Koch M, Schwarz K, Rimbach G, Hübbe P, Pan W, Sheibani-Tezerji R, Häsler R, Rosenstiel P, D'Amato M, Cloppenborg-Schmidt K, Künzel S, Laudes M, Marschall H, Lieb W, Nöthlings U, Karlsen TH, Baines JF, Franke A. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota., 2016, 48(11): 1396– 1406.
[99] Kolde R, Franzosa EA, Rahnavard G, Hall AB, Vlamakis H, Stevens C, Daly MJ, Xavier RJ, Huttenhower C. Host genetic variation and its microbiome interactions within the Human Microbiome Project., 2018, 10(1): 6.
[100] Davenport ER, Cusanovich DA, Michelini K, Barreiro LB, Ober C, Gilad Y. Genome-wide association studies of the human gut microbiota., 2015, 10(11): e0140301.
[101] Beaumont M, Goodrich JK, Jackson MA, Yet I, Davenport ER, Vieira-Silva S, Debelius J, Pallister T, Mangino M, Raes J, Knight R, Clark AG, Ley RE, Spector TD, Bell JT. Heritable components of the human fecal microbiome are associated with visceral fat., 2016, 17(1): 189.
[102] Aschard H, Laville V, Tchetgen ET, Knights D, Imhann F, Seksik P, Zaitlen N, Silverberg MS, Cosnes J, Weersma RK, Xavier R, Beaugerie L, Skurnik D, Sokol H. Genetic effects on the commensal microbiota in inflammatory bowel disease patients., 2019, 15(3): e1008018.
[103] Suzuki TA, Phifer-Rixey M, Mack KL, Sheehan MJ, Lin D, Bi K, Nachman MW. Host genetic determinants of the gut microbiota of wild mice., 2019, 28(13): 3197–3207.
[104] Crespo-Piazuelo D, Migura-Garcia L, Estellé J, Criado-Mesas L, Revilla M, Castelló A, Muñoz M, García-Casco JM, Fernández AI, Ballester M, Folch JM. Association between the pig genome and its gut microbiota composition., 2019, 9(1): 8791.
[105] Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5., 2010, 328(5975): 228–231.
[106] Salzman NH, Hung K, Haribhai D, Chu H, Karlsson- Sjöberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. Enteric defensins are essential regulators of intestinal microbial ecology., 2010, 11(1): 76–83.
[107] Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, Bridonneau C, Jegou S, Hoffmann TW, Natividad JM, Brot L, Taleb S, Couturier-Maillard A, Nion-Larmurier I, Merabtene F, Seksik P, Bourrier A, Cosnes J, Ryffel B, Beaugerie L, Launay JM, Langella P, Xavier RJ, Sokol H. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands., 2016, 22(6): 598–605.
[108] Ruan JW, Statt S, Huang CT, Tsai YT, Kuo CC, Chan HL, Liao YC, Tan TH, Kao CY. Dual-specificity phosphatase 6 deficiency regulates gut microbiome and transcriptome response against diet-induced obesity in mice., 2017, 2(2): 16220.
[109] Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T, Mujagic Z, Vila AV, Falony G, Vieira-Silva S, Wang J, Imhann F, Brandsma E, Jankipersadsing SA, Joossens M, Cenit MC, Deelen P, Swertz MA, Weersma RK, Feskens EJM, Netea MG, Gevers D, Jonkers D, Franke L, Aulchenko YS, Huttenhower C, Raes J, Hofker MH, Xavier RJ, Wijmenga C, Fu J. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity., 2016, 352(6285): 565–569.
[110] Massacci FR, Clark A, Ruet A, Lansade L, Costa M, Mach N. Inter-breed diversity and temporal dynamics of the faecal microbiota in healthy horses., 2019, doi: 10.1111/jbg.12441.
[111] van Opstal EJ, Bordenstein SR. MICROBIOME. Rethinking heritability of the microbiome., 2015, 349(6253): 1172–1173.
[112] Weissbrod O, Rothschild D, Barkan E, Segal E. Host genetics and microbiome associations through the lens of genome wide association studies., 2018, 44: 9–19.
[113] Difford GF, Lassen J, Løvendahl P. Genes and microbes, the next step in dairy cattle breeding. In: EAAP–67th Annual Meeting, Belfast, United Kingdom, 2016, 285.
[114] Espey MG. Role of oxygen gradients in shaping redox relationships between the human intestine and its microbiota., 2013, 55: 130–140.
[115] O'May GA, Reynolds N, Smith AR, Kennedy A, Macfarlane GT. Effect of pH and antibiotics on microbial overgrowth in the stomachs and duodena of patients undergoing percutaneous endoscopic gastrostomy feeding., 2005, 43(7): 3059–3065.
[116] Berry D, Stecher B, Schintlmeister A, Reichert J, Brugiroux S, Wild B, Wanek W, Richter A, Rauch I, Decker T, Loy A, Wagner M. Host-compound foraging by intestinal microbiota revealed by single-cell stable isotope probing., 2013, 110(12): 4720–4725.
[117] Tropini C, Earle KA, Huang KC, Sonnenburg JL. The gut microbiome: connecting spatial organization to function., 2017, 21(4): 433–442.
[118] Fu JY, Bonder MJ, Cenit MC, Tigchelaar EF, Maatman A, Dekens JAM, Brandsma E, Marczynska J, Imhann F, Weersma RK, Franke L, Poon TW, Xavier RJ, Gevers D, Hofker MH, Wijmenga C, Zhernakova A. The gut microbiome contributes to a substantial proportion of the variation in blood lipids., 2015, 117(9): 817–824.
[119] Talmud PJ, Hingorani AD, Cooper JA, Marmot MG, Brunner EJ, Kumari M, Kivimäki M, Humphries SE. Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: Whitehall II prospective cohort study., 2010, 340: b4838.
[120] Niu DY, Yan WL. The application of genetic risk score in genetic studies of complex human diseases., 2015, 37(12): 1204–1210.牛大彦, 严卫丽. 遗传风险评分在复杂疾病遗传学研究中的应用. 遗传, 2015, 37(12): 1204–1210.
[121] Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto J, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J. A metagenome-wide association study of gut microbiota in type 2 diabetes., 2012, 490(7418): 55–60.
[122] Wang J, Jia H. Metagenome-wide association studies: fine-mining the microbiome., 2016, 14(8): 508–522.
[123] Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, Nielsen J, Bäckhed F. Gut metagenome in European women with normal, impaired and diabetic glucose control., 2013, 498(7452): 99–103.
[124] Nielsen HB, Almeida M, Juncker A, Rasmussen S, Li J, Sunagawa S, Plichta DR, Gautier L, Pedersen AG, Le Chatelier E, Pelletier E, Bonde I, Nielsen T, Manichanh C, Arumugam M, Batto JM, Quintanilha Dos Santos MB, Blom N, Borruel N, Burgdorf KS, Boumezbeur F, Casellas F, Doré J, Dworzynski P, Guarner F, Hansen T, Hildebrand F, Kaas RS, Kennedy S, Kristiansen K, Kultima JR, Léonard P, Levenez F, Lund O, Moumen B, Le Paslier D, Pons N, Pedersen O, Prifti E, Qin J, Raes J, Sørensen S, Tap J, Tims S, Ussery DW, Yamada T, Jamet A, Mérieux A, Cultrone A, Torrejon A, Quinquis B, Brechot C, Delorme C, M'Rini C, de Vos WM, Maguin E, Varela E, Guedon E, Gwen F, Haimet F, Artiguenave F, Vandemeulebrouck G, Denariaz G, Khaci G, Blottière H, Knol J, Weissenbach J, Hylckama Vlieg VJE, Torben J, Parkhill J, Turner K, van de Guchte M, Antolin M, Rescigno M, Kleerebezem M, Derrien M, Galleron N, Sanchez N, Grarup N, Veiga P, Oozeer R, Dervyn R, Layec S, Bruls T, Winogradski Y, Erwin GZ, Renault D, Sicheritz-Ponten T, Bork P, Wang J, Brunak S, Ehrlich SD, MetaHIT C. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes., 2014, 32(8): 822–828.
[125] Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, Leonard P, Li J, Burgdorf K, Grarup N, Jørgensen T, Brandslund I, Nielsen HB, Juncker AS, Bertalan M, Levenez F, Pons N, Rasmussen S, Sunagawa S, Tap J, Tims S, Zoetendal EG, Brunak S, Clément K, Doré J, Kleerebezem M, Kristiansen K, Renault P, Sicheritz-Ponten T, de Vos WM, Zucker JD, Raes J, Hansen T, Bork P, Wang J, Ehrlich SD, Pedersen O. Richness of human gut microbiome correlates with metabolic markers., 2013, 500(7464): 541–546.
[126] Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, Amiot A, Böhm J, Brunetti F, Habermann N, Hercog R, Koch M, Luciani A, Mende DR, Schneider MA, Schrotz-King P, Tournigand C, Tran Van Nhieu J, Yamada T, Zimmermann J, Benes V, Kloor M, Ulrich CM, von Knebel Doeberitz M, Sobhani I, Bork P. Potential of fecal microbiota for early-stage detection of colorectal cancer., 2014, 10(11): 766.
[127] Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y, Lan Z, Chen B, Li Y, Zhong H, Xie H, Jie Z, Chen W, Tang S, Xu X, Wang X, Cai X, Liu S, Xia Y, Li J, Qiao X, Al-Aama J Y, Chen H, Wang L, Wu Q, Zhang F, Zheng W, Li Y, Zhang M, Luo G, Xue W, Xiao L, Li J, Chen W, Xu X, Yin Y, Yang H, Wang J, Kristiansen K, Liu L, Li T, Huang Q, Li Y, Wang J. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment., 2015, 21(8): 895–905.
The concepts and research progress: from heritability to microbiability
Chaoliang Wen, Congjiao Sun, Ning Yang
Heritability, one of the central quantitative genetic parameters, is critically important to measure the genetic variation of traits, especially in the studies of the response to selection in evolutionary biology and agriculture, and the prediction of disease risks in medicine. The statistical model and method for estimating heritability have been continually developed and improved, since the genetic variance components was first proposed by Fisher in 1918. Recently, the term “microbiability” (2), an analogous concept and estimated method to heritability, was introduced in gut microbiome research for evaluating the effect of entire microbiota on a host phenotype. In this review, we summarize the progress of statistical methods in the heritability estimation, as well as the current state of gut microbiome associations with the host genome, with a particular focus on the concept and estimated methods of microbiability. Our review will provide a reference for the future study of host phenotypic variation that can be inferred by the gut microbiota.
heritability; microbiability; association study; quantitative trait; variance component; relationship matrix
2019-05-13;
2019-09-23
国家自然科学基金项目(编号:31930105),国家现代农业(蛋鸡)产业技术体系专项资金(编号:CARS-40)和教育部长江学者及创新团队发展计划(编号:IRT_15R62)资助[Supported by the National Natural Science Foundation of China (No. 31930105), Programs for Changjiang Scholars and Innovative Research in Universities (No. IRT_15R62) and China Agriculture Research Systems (No. CARS_40)]
文超良,博士研究生,专业方向:动物遗传育种与繁殖。E-mail: clwen@cau.edu.cn
杨宁,博士,教授,研究方向:家禽分子遗传与育种。E-mail: nyang@cau.edu.cn
10.16288/j.yczz.19-130
2019/11/7 15:22:05
*:文献按发表时间从先到后排序。
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20191107.1206.004.html
(责任编委: 李辉)
猜你喜欢
中老年保健(2022年2期)2022-08-24
养猪(2022年4期)2022-08-17
中国饲料(2022年5期)2022-04-26
科学(2020年4期)2020-11-26
科学(2020年3期)2020-11-26
当代水产(2020年3期)2020-06-15
湖北畜牧兽医(2015年11期)2016-01-11
湖北畜牧兽医(2015年11期)2016-01-11
小星星·阅读100分(高年级)(2015年11期)2015-11-28
医学研究杂志(2015年12期)2015-06-10
