
我国药品专利强制许可制度分类实施探讨
2019-11-23 14:38陈学宇
华侨大学学报·哲学社会科学版 2019年5期
关键词:专利法
摘 要:药品专利强制许可制度基于平衡药品专利权和公共健康利益而生,是世界各国在TRIPS协定框架下保障仿制药供应、降低药品价格和应对公共健康危机的重要制度供给。在经历三次修订后,我国专利法现已建立了药品专利强制许可制度的主体框架,构造了药品专利强制许可一般制度和药品出口专利强制许可特殊制度两类制度安排,设计了五种实施路径,并初步建立了保障实施的配套制度。法律制度障碍目前仍是阻碍我国药品专利强制许可制度实施的重要因素,主要表现为药品专利强制许可一般制度的可操作性不强、药品出口专利强制许可特殊制度的内容和功能设计不尽合理,以及缺乏部门协作与经济激励等配套制度。建议我国修正《专利法》中专利强制许可章节的结构与条款,完善《专利法实施细则》等行政法规和部门规章的相关内容,并尽快健全基于药品专利强制许可制度的部门协作机制。
关键词:药品专利;强制许可制度;专利法;仿制药
作者简介:陈学宇,厦门大学知识产权研究院博士研究生,主要研究方向:知识产权法、药事法和竞争法。(Email:chxuey2006@163.com;福建 廈门 361005)。
基金项目:国家知识产权局横向课题“出口等三种情形下专利强制许可程序实务研究”(k8215008)
中图分类号:D923.42文献标识码:A
文章编号:1006-1398(2019)05-0099-12
一 引 言
药品专利强制许可制度是平衡药品专利权人利益和公共健康利益的一种重要手段,是《与贸易有关的知识产权协定》(以下简称TRIPS协定)框架下世界贸易组织(以下简称WTO)各成员国专利法律制度的基本组成部分。进入21世纪以来,药品专利强制许可制度的有效实施日益成为广大发展中国家和最不发达国家保障仿制药供应、降低药品价格和应对公共健康危机的重要制度供给。(WTO,WIPO,WHO,Promoting Access to Medical Technologies and Innovation:Intersections between Public Health,Intellectual Property and Trade.WTO ISBN 978-92-870-3964-4,2013.)
例如与我国同为人口众多的发展中国家的印度,虽然迟至2002年才引入了药品专利强制许可制度,但已于2012年实施了首个药品专利强制许可,极大的减轻了相关民众的用药经济负担。
据统计,2001年至2016年间,相关国家和地区总共实施了48件药品专利强制许可,取得了广泛的经济和社会效益。(Ellen FM‘t Hoen,Jacquelyn Veraldi,Brigit Toebes, Medicine Procurement and The Use of Flexibilities in The Agreement on Trade-Related Aspects of Intellectual Property Rights,2001-2016.Bull World Health Organ,Vol.3,2018,pp.185-193.)我国早在1984年第一部《专利法》制定之时,就已认识到了专利实施强制许可制度的重要性;后续受美国等国和WTO的影响,我国《专利法》在1992年、2000年以及2008年修订过程中,在授予和不断强化药品专利保护的同时,对药品专利强制许可制度进行了系统性地构造和完善。经过这三次专利法修订及后续的行政法规、部门规章的调整,我国已构建了全面符合TRIPS协定要求的药品专利强制许可制度框架。随着2013年以来陆勇案、魏则西案等药品可及性案件的相继发生以及2018年《我不是药神》等现象级电影的热映,我国政府、学术界和民间对“药品专利强制许可制度的实施问题”的探讨日益广泛而深入,启动药品专利强制许可制度的社会环境也逐步形成。在此背景下,国务院办公厅于2018年4月3日印发了《关于改革完善仿制药供应保障及使用政策的意见》(以下简称《意见》),提出要“明确药品专利实施强制许可路径”,并将依法分类实施药品专利强制许可,以期提高我国仿制药品的供应保障能力。
我国的药品专利强制许可制度自1984年建立以来就一直处于“零实施”的状态,(对此问题学术界实际上存在一些争议。自2008年《专利法》完成第三次修改后,我国也曾出现了一例类似申请药品专利强制许可的尝试,即白云山版“达菲”案件:2009年11月16日,广州白云山制药总厂公告称其已向国家药监部门提交了“提前受理我厂仿制磷酸奥司他韦原料及胶囊的注册申请”,希望提前批准开展生物等效性试验,但该尝试最终未能成功。该事件被一些学者认为是我国实施强制许可的重要尝试,参见袁泉、邵蓉:《从白云山版“达菲”事件看我国药品专利强制许可制度》,《中国新药杂志》2010年第16期,第1392—1395页。但本文对此持不同观点,因为该尝试主要涉及药品监管部门的药品就注册活动和《专利法》第69条第5项Bolar例外的适用,而非药品专利强制许可制度实践。)究其原因,必然涉及政治、经济、法律等诸多层面的问题。例如从医药产业的发展层面上看,我国仿制药产业发展水平与制药业发达国家仍存在较大差距,国内公众和医院对仿制药质量和疗效的信心不高,(黄智然、刘诗洋、魏晓晶等:《北京市85家二、三级公立医院心血管类原研药与仿制药利用分析》,《药物流行病学杂志》2017年第7期,第490—495页。)一定程度上影响了医药产业界的实施意愿。但从学术界针对药品专利强制许可制度的专门性研究成果来看,法律制度层面的不完善仍然是限制我国药品专利强制许可制度实施的重要因素。例如,现行药品专利强制许可制度设计仍未充分考虑我国国情,很大程度上只是对TRIPS规定的复制,甚至存在高于TRIPS协定标准的问题。(郝敏:《药品专利强制许可制度在发展中国家的应用——从“抗癌药代购第一人”陆勇事件谈起》[JP5],《知识产权》2015年第8期,第95—101页。)现行专利法尚缺乏对药品相关“公共健康”概念的定义,导致相关制度的可操作性较差,进而也限制了药品专利强制许可制度的有效实施。(刘立春、朱雪忠:《与药品专利强制许可相关的“公共健康”含义》,《中国卫生经济》2015年第2期,第73—78页。)药品专利强制许可制度存在实施条件苛刻、程序繁琐等问题,不利于申请人的操作,降低了申请人的申请积极性。(张晓敏:《公众健康权与药品专利权:利益冲突及其政策选择》,《财经科学》2011年第8期,第103—109页。)此外,缺乏对申请人动因缺陷的补救措施、药品专利强制许可补偿数额的不确定性、缺乏建立强制许可药品的质量监督与救济机制和药品强制许可程序期限过长等原因,也对药品专利强制许可制度的实施形成了制度障碍。(赵利:《我国药品专利强制许可制度探析》,《政法论坛》2017年第2期,第146—151页。)
还有学者借鉴印度等仿制药发达国家的经验,提出我国应当及时出台药品专利强制许可实施细则和适当放宽药品专利强制许可申请理由等内容。(陈永法、雷媛、伍琳:《印度药品专利强制许可制度研究》,《价格理论与实践》2018年第8期,第90—93页。)
然而纵观我国学术界相关研究,虽然药品专利强制许可制度仍有待完善已是共识,但是从分类实施角度对不同类型的药品专利强制许可制度进行研究的相关成果尚不多见,并且从药品专利强制许可实施全程管理的角度分析强制许可需要的专利法以外的配套制度资源问题(例如药品监管、研发资助等等)也较为缺乏。这既反映了我国药品专利强制许可制度研究还存在深入拓展的理论空间,也与《意见》提出的促进分类实施的现实需要存在距离。
当下我国药品专利强制许可制度的相关研究方向正从制度构造层面转向制度实施层面,而研究的核心旨趣也已转向推動药品专利强制许可的具体分类实施问题,以期尽快实现其保障相关仿制药供应和维护公共健康的制度初衷。为因应这一趋势,本文将从分类实施和全程管理的基本理念出发,分析我国专利法上药品专利强制许可制度的现行制度构造,探讨实施药品专利强制许可制度面临的主要制度障碍,最后有针对性地提出完善建议,希冀对打破药品专利强制许可制度“零实施”状态有所助益。
二 我国药品专利强制许可制度分类实施的制度构造
(一)两项制度安排
在TRIPS协定制定之初,当时发达国家主导下的TRIPS协定仅在第31条中制定了专利强制许可一般制度。由于TRIPS协定要求各成员国对药品实施普遍的专利保护,并对药品专利强制许可制度的实施设定了严苛的限制条件,忽略了发展中国家和最不发达国家的公共健康状况和药品生产供应能力,曾一度引发了印度为代表的发展中国家的普遍不满。在之后的TRIPS协定实施过程中,跨国制药企业凭借药品专利向发展中国家索取高价和限制供应,甚至还引发了南非等国发生了药品专利权与公共健康的激烈冲突事件。(Ellen‘t Hoen,Jonathan Berger, Alexandra Calmy,Driving a Decade of Change:HIV/AIDS,Patents and Access to Medicines for All.Journal of the International AIDS Society,Vol.14,2011,p.15.)一系列争议的发生,迫使WTO不得不正视第31条药品专利强制许可一般制度存在的严重问题。为减轻发展中国家和最不发达国家履行TRIPS协定的公共健康负担,帮助这些国家在应对公共健康危机时更方便地获得相应的仿制药品,WTO于2001年至2005年间相继通过了《TRIPS协定与公共健康的宣言》(以下简称《多哈宣言》)、《关于实施TRIPS协定与公共健康多哈宣言第六段的决议》(以下简称《总理事会决议》)和《修改TRIPS协定议定书》(以下简称《议定书》)。“《多哈宣言》三部曲”不仅对药品专利强制许可一般制度进行了更为灵活的解释,而且在TRIPS第31条下特别增加了第二部分,构建了药品出口专利强制许可特殊制度。(陈婷、林秀芹:《<多哈宣言>实施中的法律障碍及发展前景展望——<多哈宣言>实施效果评估》,《国际经济法学刊》2013年第2期,第188—208页。)至此,TRIPS协定第31条形成了两套制度安排:其一是药品专利强制许可一般制度,令具有相关药品生产供应能力和渠道国家,有机会以本地生产或者进口的方式实施药品专利强制许可制度;其二是药品出口专利强制许可特殊制度,使缺乏相关药品生产供应能力和渠道的国家,有机会通过协调进口强制许可药品的方式保障仿制药的有效供应。
回顾我国药品专利强制许可制度的演进历程,虽然我国早在第一部《专利法》时就已设计了专利强制许可制度的宽泛性规定,但内容十分简单。而现行药品专利强制许可制度实际上是后续为适应TRIPS协定第31条的要求而进行系统性修正的结果,(林秀芹:《中国专利强制许可制度的完善》,《法学研究》2006年第6期,第30—38页。)与1984年的相关规定已相去甚远。因此,可以认为我国药品专利强制许可制度的主要渊源来自于TRIPS协定第31条。参考TRIPS协定第31条的发展过程和设计思路,本文认为,我国药品专利强制许可制度亦可大致分为药品专利强制许可一般制度和药品出口专利强制许可特殊制度两项制度安排:
第一,药品专利强制许可一般制度。我国专利法上的药品专利强制许可一般制度是指国家专利行政机关依据法定条件和程序,不经药品专利权人的同意,向特定对象颁发实施该专利的许可,并由被许可人向药品专利权人支付使用费的制度。从启动事由来看,我国药品专利强制许可一般制度还可以进一步细分为涉及未实施或未充分实施专利的强制许可,涉及垄断行为的强制许可,涉及国家出现紧急状态、非常情况和为了公共利益目的的强制许可和从属专利的强制许可四种类型。药品专利强制许可一般制度的制度价值主要体现在保障我国国内的仿制药供应需求方面,因此主要以满足本国国民的药品消费要求为限,一般不涉及出口问题,(涉及垄断情形的强制许可范围可能存在特例。因为该情形下药品专利强制许可的颁布需以消除药品专利垄断对国内的不利影响为目的,鉴于该影响可能来自国外,故强制许可的范围也不应限制在国内。)但可能涉及药品进口问题。即如果该药品在出口国不受专利保护,那么我国只需按照药品专利强制许可一般制度,颁布允许被许可人进口该药品的强制许可;如果该药品在出口国也受专利保护,那么我国就必须借助TRIPS协议第31条之二规定的药品出口专利强制许可特殊制度,向WTO理事会履行通报手续,同时协调出口国按照该制度颁发强制许可并履行通报手续。(国家知识产权局条法司编:《<专利法>及<专利法实施细则>第三次修改专题研究报告》,北京:知识产权出版社,2006年。)
第二,药品出口专利强制许可特殊制度。药品出口专利强制许可特殊制度是指国家专利行政机关为了公共健康目的,不经药品专利权人的同意,对取得专利权的药品授予制造并将其出口到符合中国参加的有关国际条约规定的国家或者地区的强制许可,不涉及药品进口问题。我国药品出口专利强制许可制度的特殊性表现在:其一,该制度定位在于协助他国解决药品可及性问题和改善公共健康状况,其二,强制许可的内容仅限于对专利药品的制造和出口,并且只能出口到符合中国参加的有关国际条约规定的国家或者地区,不能对国内药品市场产生影响。药品专利强制许可一般制度与药品出口专利强制许可特殊制度的分别安排,反映了药品专利强制许可制度功能的不同侧面,在价值目标上紧密互补,共同服务于我国和世界的仿制药供应和公共健康状况之改善。
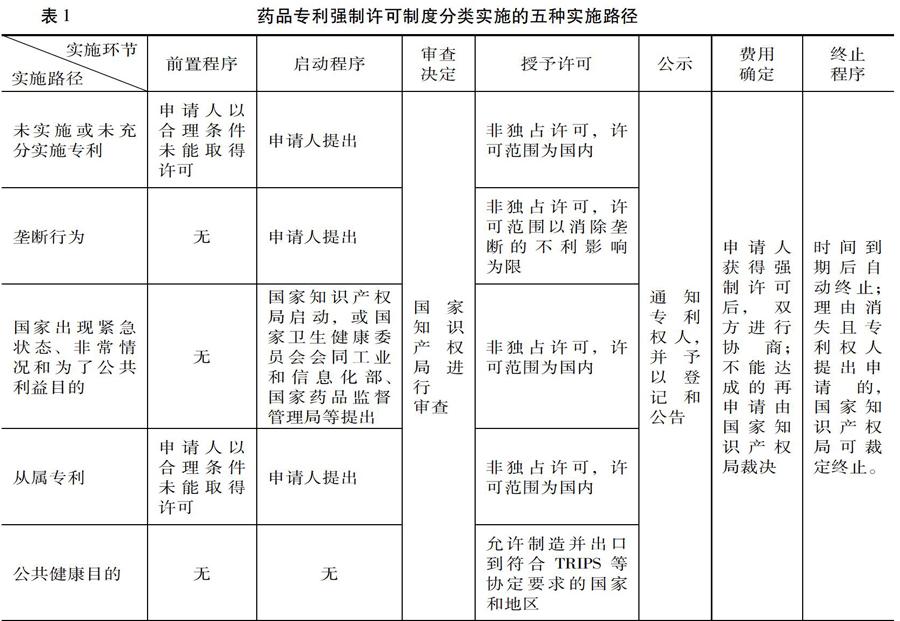
(二)五类实施路径
药品专利强制许可制度的实施情形具有多元性,不同的强制许可类型意味着不同的实施路径构造。(详见表1)
药品专利强制许可一般制度的四种实施路径。专利法上的药品专利强制许可一般制度的四种路径的实施均涉及启动、审查、授予、公示、费用确定和终止六个环节。但在未实施或未充分实施专利和从属专利的两种情形下,专利法还对申请人提出申请设置相应前置程序(即对于必须是申请人与专利权人进行先行协商,申请人有提出合理的条件,但在合理期限内未获得许可)。对涉及国家出现紧急状态、非常情况或者为了公共利益目的的强制许可类型,一般而言无须申请人提出,国家知识产权局可直接自行启动,但为了推动有效实施,2018年《意见》在该情形下还对其启动程序进行了完善(详见后文)。
药品出口专利强制许可特殊制度的实施路径。药品出口专利强制许可特殊制度的实施也包括启动、审查、授予许可、公示、费用确定和终止六个环节,除启动事由(公共健康目的)和授 予强制许可的内容(制造并出口到相关国家)与药品专利强制许可一般制度存在差异外,其审查、公示、费用确定和终止四个环节均可适用药品专利强制许可一般制度的相关规定。但针对该制度是否应设置前置程序以及该制度的启动程序如何等问题,现行《专利法》尚无明确规定。
(三)实施配套制度
纵观各国的相关法治实践,药品专利强制许可制度的实施从来不只是专利法领域的问题,其实施全过程必然涉及与卫生健康、药品监管、药品创新等诸多方面的制度衔接。实际上,随着世界医药产业的发展,药品知识产权与卫生健康、药品监管、贸易政策等领域之间的密切联系和制度交融现象日益凸显,不同领域之间的制度互动与协调机制正在不断完善,对药品专利强制许可等制度的实施将产生更加深刻影响。(陈学宇:《<跨太平洋伙伴关系协定>中涉药品监管的知识产权条款研究》,《中国药学杂志》2017年第6期,第513—518页。)例如,随着各国对仿制药质量安全的关注,药品监管部门对仿制药上市的要求一直在不断提高。在获得强制许可后,强制许可药品作为仿制药,不仅在上市前必须通过一系列的药品安全性、有效性和质量可控性审评审批,而且在上市后还必须应对生产质量控制、药品销售和医疗保险等制度的制约,造成了药品仿制难度加大、生产成本提升和强制许可药品销售难度增加等问题,直接影响着药品专利强制许可制度实施的有效性。因此,药品专利强制许可制度的相关研究也必须从多领域融合的视角进行考量,不仅专利法需要给予药品专利强制许可制度足够的重视,而且卫生健康部门、药品监管部门、科技部门等也必须进行协同运作,方能更为全面地保障我国药品专利强制许可制度的顺利实施。
近期我国已开始了基于实施药品专利强制许可的配套制度构建尝试,正在覆盖该制度实施的更多环节。如在2017年10月中共中央办公厅和国务院办公厅联合发布的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(简称《审评审批制度改革意见》)中,强调“要建立专利强制许可药品优先审评审批制度”,并要求原“国家卫生计生委会”会同有关部门规定公共健康受到重大威胁的情形和启动强制许可的程序。此外,为进一步推动药品专利强制许可制度精准实施,2018年国务院办公厅印发的《意见》从部门协作的角度也增加了该制度启动的另一种可能,作为国家出现紧急状态、非常情况和为了公共利益的目的强制许可的前置程序。即在国家出现重特大传染病疫情及其他突发公共卫生事件或防治重特大疾病药品出现短缺,对公共卫生安全或公共健康造成严重威胁等非常情况时,由国家卫生健康委员会会同工业和信息化部、国家药品监督管理局等部门向国家专利行政部门提出启动建议。
三 我国药品专利强制许可制度分类实施的主要制度障碍
(一)药品专利强制许可一般制度的可操作性不强
1.申请人条件高于TRIPS协定设定之标准
我国专利法对涉及未实施或未充分实施专利和垄断行为两种类型的申请人设置了明显高于TRIPS协定相关要求的申请条件,因而被普遍認为是一项TRIPS-plus标准。(TRIPS-Plus标准是指超过TRIPS协定要求的法律标准,主要体现在对TRIPS协定中包括第31条在内的灵活性条款进行限制。)在TRIPS协定中的强制许可申请人被称为“拟使用人”,并未被设置申请条件;实践中,印度等国也认为,只要是“利益相关人”就可以提出申请。(郝敏:《药品专利强制许可制度在发展中国家的应用——从“抗癌药代购第一人”陆勇事件谈起》,《知识产权》2015年第8期,第95—101页。)但是,我国《专利法》及相关规范性文件均要求,涉及垄断和未(充分)实施专利的强制许可申请人应当“具备实施条件”。虽然对申请人设定“具备实施条件”的要求或可以在一定程度上保障强制许可的有效实施,避免强制许可资源的浪费,但是要求药品专利强制许可申请人在一开始就完全具备相应的实施条件实际上并不合理。一方面,仿制药企业提前筹备相应的实施条件可能面临着侵犯药品专利权的法律风险;另一方面,医药产业是技术密集型产业,由于被仿制专利药往往都是技术含量较高的药品,对相应的资金和技术要求较高;在难以确定是否能够获得强制许可的情况下,仿制药企业就投入巨大成本和准备完整的实施条件的决策也并不理性。申请门槛过高,是实施药品专利强制许可的直接阻碍。
2.紧急情况、非常情况和公共利益目的等核心概念难以把握
我国现行《专利法》的第49条明确规定了“紧急情况”“非常情况”“公共利益目的”是国家知识产权局实施强制许可的事由之一,可实施的具体情形包括防治环境污染目的等(全国人大常委会法制工作委员会经济法室编著:《<中华人民共和国专利法>释解及实用指南》,北京:中国民主法制出版社,2012年,第118页。)(第50条规定的是“公共健康目的”,是“公共利益目的”在公共卫生领域的具体诉求)。但是因为“紧急情况”“非常情况”“公共利益目的”概念本身具有模糊性的特点,难以为申请人和审查部门所把握。虽然2018年《意见》对上述进行了一定程度的补充说明,但仍然十分模糊。(《意见》第十二项所认为的紧急状态,是指在国家出现重特大传染病疫情及其他突发公共卫生事件或防治重特大疾病药品出现短缺,对公共卫生安全或公共健康造成严重威胁等非常情况。)如果是由艾滋病等传染病引发的公共健康危机,我国尚可勉强援引《多哈宣言》(《多哈宣言》第5条第C项便已明确列举了“艾滋病、结核病、疟疾以及其他传染病”这些疾病范围,如果各国为应对这些疾病引发的公共危机颁发强制许可,当然是符合“公共利益目的”和“公共健康目的”。)和《意见》第十二项进行补充解释;但如果是由癌症等非传染性疾病引发的仿制药可及性问题,相关概念的内涵就容易引起诸多争议。核心概念的难以把握会进一步导致药品专利强制许可的申请和实施结果难以预测,削弱申请人的积极性。此外,国家知识产权局也尚未对相应概念形成明确的审查标准,对于审查和授予强制许可的准确性和时效性也存在不利的影响。
3.专利使用费的确定程序相对繁琐且缺乏计算标准
现行专利法对药品专利强制许可使用费的规定在程序和实体两个层面均存在问题。在药品专利强制许可情形下,被许可人仍应当向专利权人支付相应的使用费作为补偿,这是强制许可制度的基本要求。但是从程序法上看,按照我国《专利法》《专利法实施细则》和《专利强制许可实施办法》规定,国家知识产权局对强制许可的审查与专利使用费的审查实行的是“两裁分离”程序,使得申请人在获得强制许可后还必须再与专利权人行协商、向政府申请乃至进行审查后,方能确定使用费用,明显增加了申请人执行强制许可的机会成本。从实体法上看,《专利法》《专利法实施细则》和《专利实施强制许可办法》也均未对使用费的计算标准进行规定。作为专利许可实践中最容易引起争议和最难以达成协议的核心问题,未规定计算标准,便不能给予申请人提供合理的经济预期,也会增加当事人发生争议的可能。
4.强制许可的时间期限难以判断且不能延长
我国药品专利强制许可的期限采取的是有限制且可以提前终止的设计思路。药品专利强制许可的时间期限也是我国国家知识产权局颁发强制许可时应确定的重要事项,但现行《专利法》仅在第55条第2款对强制许可的时间期限进行了简单规定。仅说明了强制许可的时间期限由国家知识产权局依个案的具体情况审查决定,但确定时间期限的判断标准并不清楚。同时,强制许可的期限除届满自动终止外,还可能会因为专利的有效性、强制许可理由不再存在而被提前终止。可见,关于强制许可期限的制度设计不仅缺乏判断标准,让人难以把握,而且仅规定强制许可事由提前终止、而无视事由可能持续的情况无疑显得过于乐观,可能面临同一事件引发的强制许可必须进行多次申请、审查和颁布的问题。
(二)药品出口专利强制许可特殊制度的内容和功能不尽合理
1.制度内容过于简单,缺乏前置程序和启动主体等规定
在《专利法》第三次修订之时,我国对药品专利出口强制许可的特殊性并未进行深入研究,而现行《专利法》也仅在第50条和第52条对其进行了相应的规定,制度内容设计过于简单,难以适应该制度的实践需要。实际上,按照“《多哈宣言》三部曲”,该制度的实施需要在WTO框架下,由进口国、出口国两个法域内的政府部门和企业等多主体共同努力,且必须经过进口国通知WTO和相关政府或者申请人向我国政府提出申请等环节作为前置程序,涉及国家知识产权局对外国政府(或其代理人)出口诉求的接收和启动,实施流程和执行程序较为繁琐。因此,药品出口专利强制许可特殊制度的实施,除了可以适用药品专利强制许可一般制度的审查、公示、费用确定和终止等环节的规定外,至少还必须对该制度的前置程序和启动程序问题进行专门的规定。
2.出口范围过窄抑制被许可人的实施积极性
经过多年的发展,我国医药产业发展水平整体有了质的飞跃,且在部分领域的药品创新和仿制已达到较高的水平和可供出口的程度,可以为世界公共健康状况做出更大的贡献。(例如青蒿素等自主研发的产品为疟疾等传染性疾病的防治做出巨大貢献;中国的PD-1单抗等抗肿瘤药物屡屡卖出天价,标志着我国相关领域的生物制品领域的研发能力得到了更多的国际认可;2015年,我国自主研发的全球首个2014基因突变型埃博拉疫苗获得了世界卫生组织(WHO)、西非国家和国际社会的一致好评等。)然而,现行《专利法》第50条的规定对药品出口专利强制许可的出口范围进行了限定,即国家知识产权局可以给予制造并将其出口的范围仅是符合中华人民共和国参加的有关国际条约规定的国家或者地区。该条款的范围限定主要是遵守TRIPS协定有关规定的产物;根据《议定书》附件中关于“有资格进口的成员”的解释,主要是指任何最不发达成员及任何已向WTO理事会通报意图作为进口成员利用药品出口专利强制许可特殊制度的其他成员。实际上,由于进口国的经济发展水平往往比较低,其需求量很可能不足以让仿制药企业的生产达到规模效应水平,相关药品的平均生产成本比较高,(《议定书》附件一的第3段规定了解决出口量过小的一个解决方案,即区域性的联合订购,但条件仍然十分严苛。)导致仿制药企业不仅缺乏适当获利的可能性,甚至可能无法收回研发和生产的成本,自然缺乏积极性。
(三)部门协作和经济激励等配套制度有待完善
1.实施药品专利强制许可的部门协作机制尚不健全
如前所述,在国家知识产权局颁发强制许可后,仿制药的有效生产和使用便需要依靠国家知识产权局与其他部门的高效对接与协作。但目前我国涉及药品强制许可的部门协作机制还远未完善,主要问题包括:其一,相关规定过于分散,缺乏覆盖药品专利强制许可制度实施全过程的统筹安排。目前我国对药品专利强制许可制度的规定主要集中在《专利法》《专利法实施细则》和《专利实施强制许可办法》,但涉及部门协作的规定则分散在由中共中央办公厅和国务院办公厅发布的若干规范性文件之中。这些规定从实施环节的角度进行了职能划分,但缺乏从药品专利强制许可制度实施全过程的视角进行统一安排。其二,仅有原则性内容,缺乏可操作性。涉及药品强制许可部门协作机制的规定,目前仅有《审评审批制度改革意见》第三部分第14条和《意见》第三部分第12条,均属于原则性规定,尚未见相关部门出台针对性的细则或者指南。
参考国际实施经验,药品专利强制许可制度的有效实施,也需要对药品管理法等领域进行相应的调整。例如,作为实施药品专利强制许可制度经验较为丰富的国家之一,加拿大为了推动药品出口强制许可特殊制度的实施,于2004年专门通过了《加拿大药品获取法》,不仅修改了加拿大专利法,还系统性地调整了食品药品法的相关内容。该法案包含19部分100多项条款,对授权部门、药品范围、质量安全等等均进行了详细的规定,以适应实践的要求。(熊建军:《
2.缺乏对仿制企业提供必要的经济激励
现行药品专利强制许可制度缺乏对仿制药企业的必要经济激励也是一个值得反思的问题。如果要实施药品专利强制许可制度,我国仿制药企业就必须承担强制许可的申请、审查和药品注册程序引起的程序性执行成本,同时还必须承担研发、生产和销售带来的一系列的经济成本压力。主要体现在:第一,药品研究、开发和制造成本。如前所述,我国制药产业在抗艾滋病、癌症等领域的研发和仿制水平原本就有待提高,而涉及这些公共健康问题的专利药品多对技术水平和设备有相当高的要求,国内企业要想成功仿制往往需组建高水平的研发团队和购买价格昂贵的技术设备,并经一系列研究和试验才能仿制成功,加之时间紧迫,药品仿制的成本要远高于一般仿制药。第二,专利使用费。根据TRIPS和专利法的要求,被许可人仍然要支付一定的专利使用费,国际经验多为销售额的0.5%~6%之间。(印度在2012年的Sorafenib案件中支付了销售额的6%(6%为印度强制许可使用费的上限);欧盟在紧急状态下的强制许可费用为合同价值的5%;加拿大的幅度为0.02%~3.5%之间;泰国的许可使用费率最低,例如在2007年授予的Lopinavir+Ritonavir案件中,仅支付了销售总额的0.5%作为补偿。)虽然该比例不高,由于仿制药的价格较低和利润空间有限,专利使用费也是一项不容忽视的成本。第三,药品营销成本。在药品专利强制许可情形下,被许可人还必须采用特定的标签或者标记注明“该药品是依据强制许可而制造的产品”,而这极可能导致患者和医生在购买和使用过程中存在误解,进而影响该药品的销售。(此外,前文提及的出口范围过窄问题对此亦对仿制药企业存在不利影响,在此不再赘述。)
作为一项高经济投入、低经济回报的举措,虽然药品专利强制许可制度的实施展现了国内仿制药企业的高度社会责任感,但我国政府和法律制度也应当对其提供一定的经济激励,分担一定的执行成本。毕竟,对被许可人的合理商业利益进行考虑也是维持企业积极性的必要措施,否则一旦剥夺其获得合理补偿及适当投资回报的可能性,甚至可能导致有能力的仿制药企业自始就缺乏实施的动力。(Jennifer A,Lazo,The Life-saving Medicines Export Act:Why The Proposed U.S.Compulsory Licensing Scheme Will Fail To Export Any Medicines Or Save Any Lives.Brooklyn Journal of International Law,Vol.2,2007,pp.238-276.)而且各国的实践也表明,无论是制药公司可能多么无私,或有多么严重的流行病如艾滋病,如果没有一定的经济保障,有关药品专利强制许可的法律对改善生命或死亡问题作用也将是有限的,民众依然可能难以获得基本药物。([JP5][德]Reto Hilty:《专利保护宣言——TRIPS协定下的规制主权》,张文韬、肖冰译,林秀芹校,《中外知识产权评论》2015年第1卷,第19页。)
四 完善我国药品专利强制许可制度的若干建议
(一)修正《专利法》上专利实施强制许可制度的结构与条款
1.实现强制许可裁决与使用费裁决的“两裁合一”
考虑到实践中当事人往往难以就强制许可使用费问题达成一致,要求双方在许可颁发之后再行协商的规定,增加了双方的不必要负担。因此,我国应当参考加拿大、印度、泰国等国的强制许可立法和实践经验,实现强制许可裁决与使用费裁决程序上合一,在强制许可审查决定时就专利使用费一并进行审查决定。故建议将《专利法》第57条第二句“付給使用费的,其数额由双方协商;双方不能达成协议的,由国家知识产权局裁决。”修改为:“申请人在提交强制许可申请时,应同时提出强制许可使用费数额或计算标准,由国家知识产权局一并裁决”。
2.单独设立“药品出口专利强制许可特殊制度”一节
一方面,考虑到药品出口强制许可是不同于一般强制许可的特殊制度安排,我国应当参考TRIPS和加拿大的立法经验,将涉及药品出口强制许可的特殊条款集中,独立成节。易言之,即在现行《专利法》第六章“专利实施的强制许可”应分为“专利强制许可的一般制度”与“药品专利出口强制许可的特殊制度”两节,各自独立。另一方面,在“药品出口专利强制许可特殊制度”一节中,应当首先增加前置程序和启动条件等规定。在前置程序方面,如果我国是以出口为目的,应当以收到他国的药品出口需求照会为前提;如果我国是以进口为目的,则必须向WTO进行相应的通知。在启动条件方面,国家知识产权局应当以在收到我国外交部门的正式通知或他国政府(代理人)的申请后方能启动。同时,鉴于加拿大等国的在药品出口专利强制许可特殊制度方面存在程序极其繁琐等问题,不利于该制度的很好实施,(熊建军:《
3.扩大药品出口专利强制许可特殊制度的实施范围
实践中,欧盟及其成员国、印度、菲律宾、中国台湾等40个国家和地区选择将出口范围扩大至非WTO成员国,其中印度等国已将强制许可药品的出口范围扩张到了任何“不具备相应药品生产能力的国家和地区”。(Roger Kampf,Special Compulsory Licences for Export of Medicines:Key Features of WTO Members implementing Legislation.WTO Staff Working PaperERSD-2015-0731,2015.)这一制度设计,既符合《多哈宣言》等TRIPS协定相关文件所确立的目标和原则,还能够进一步提升药品出口专利强制许可特殊制度的生命力和企业的开展积极性。参考上述国家的经验,我国《专利法》第50条可以将该出口范围扩大至“任何不具备相应药品生产能力的国家和地区”,方便仿制药企业根据各国公共健康现实状况的需要开展实践。
(二)完善《专利法实施细则》等行政法规和部门规章相关内容
1.扩张性解释“具备实施条件”概念
关于现行专利法对申请人必须具备实施条件的标准过高的问题,有学者认为,具备实施条件“可以作为专利主管部门审核时决定是否给予批准的考察因素,而不宜作为申请主体的资格限制条件”(张冲、叶红兵:《论我国专利强制许可制度的改革与完善》,《科技管理研究》2013年第17期,第167—170+176页。)。但该思路仍要求国家知识产权局承担起国家药品监督管理局的审查职责,既超越了国家知识产权局的能力范围,也容易导致强制许可的审查时间过长的问题,因此也不可取。本文认为,完全取消具备实施条件的限制可能导致强制许可申请的泛滥和强制许可审查资源的浪费,但要求申请人在申请之时就完全具备所有的实施条件也并不合理。因此宜对“具备实施条件”采用扩张性解释,要求申请人应当在申请时其已初步具备实施条件即可;但申请人必须在申请时作出承诺和提供证据,保证其将在获得药品专利强制许可后的一定时间内获得相关药品生产制造和销售的全部条件;而国家知识产权局还可以与国家药品监督管理局进行沟通确认,并请其对申请人的后续生产和销售活动进行严格监管,并及时通报申请的相关信息。
2.明确专利使用费的考量因素和计算标准
《专利法》上关于专利使用费的规定亦存在进一步解释的空间,可在《专利法实施细则》《专利实施强制许可办法》中增加使用费的实体计算标准相关规定。在《专利法實施细则》中,可借鉴国际上的强制许可审查经验,规定专利强制许可使用费的确定,应当综合考虑自愿情况下的专利许可费、研发成本、专利的经济价值、强制许可的收益、损害赔偿情况及专利的合理奖励等因素,进行综合裁量。(林秀芹:《TRIPs体制下的专利强制许可制度研究》,北京:法律出版社,2006年,第342—348页。)在后续《专利实施强制许可办法》中,可参考2001年联合国发展报告提出的药品专利强制许可补偿费计算方式,(朱怀祖:《药品专利强制许可研究》,北京:知识产权出版社,2011年,第227页。)明确最终的使用费幅度。根据该计算方法,可以将仿制药销售金额的4%作为药品专利强制许可使用费的基准点,并在此基础上可以综合考虑各种因素上下浮动2%。
3.形成标准较为明确且可延长的许可期限
从国际立法和执法情况来看,大部分国家都有关于专利强制许可期限的具体考量标准,包括依申请人的申请、专利有效期以及强制许可理由等。同时,关于专利强制许可期限的调整,各国也存在一些灵活的规定,既可能缩短,也可能延长。如加拿大便设定了可延展的初始有效期限制度,欧盟的塞尔维亚、马其顿则设立了有效期再调整程序,等等。(Roger Kampf,Special Compulsory Licences for Export of Medicines:Key Features of WTO Members implementing Legislation.WTO Staff Working Paper ERSD-2015-0731,2015.)参考国际经验,我国亦可以在《专利法实施细则》或《专利实施强制许可办法》中明确如下内容。其一,确定药品专利强制许可的有效期限,应当以申请人的主张为基础,结合强制许可理由的严重程度、专利有效期、药品注册时限等因素进行综合考量。其二,强制许可的时间到期但强制许可的事由仍然存在的,国家知识产权局可以根据被许可人的请求,经审查后直接作出在一定时期内继续实施强制许可的决定。
(三)健全基于药品专利强制许可制度的部门协作机制
1.联合制定发布药品专利强制许可制度的实施指南及其附件
随着适时实施药品专利强制许可制度已成为国务院各相关部门的共识,当前有关规定分散在不同的政策文件中的现状明显不利于该制度的实施,必须从覆盖药品专利强制许可制度实施全过程视角进行统一调整。对此,已有学者建议,国家有关部门应当及时整合相关政策文件中有关专利强制许可的规定,“出台全新的覆盖专利强制许可全过程的实施细则”
(陈永法、雷媛、伍琳:《印度药品专利强制许可制度研究》,《价格理论与实践》2018年第8期,第90—93页。)。本文认为,目前我国对药品强制许可制度的实施主要涉及国家知识产权局、国家卫生健康委员会、国家药品监督管理局、科技部和国家医疗保障局等部门,各部门既应当制定统一的、覆盖实施全过程的药品专利强制许可实施指南,也应当在各自职能范围内制定相应环节的配套文件:其一,可采用“会同”立法的方式共同制定并发布实施指南,以提升立法和实施的效率;其二,各部门还应该根据本部门的职能内容制定相应的目录、意见或者规定,作为统一实施指南的附件,确保各部门内部的流程运行顺畅。
2.制定符合我国公共健康状况的可强制许可药品指导目录
从实用主义的角度出发,为方便药品专利强制许可制度实施,借鉴加拿大的药品强制许可目录经验,国家卫生健康委员会和国家药品监督管理局可以从我国当前的疾病谱和医药产业发展现状出发,制定可授予药品专利强制许可的药品品种目录,作为实施指南的重要附件之一。该目录应当尽可能广泛地包含解决我国和其他发展中国家公共健康问题所涉及的医药产品。一方面,药品目录包括的可被授予药品专利强制许可的药品应扩展到“医药产品”的范围,即应包括我国《药品管理法》意义上的药品,也应当包括医疗器械、药械组合等其他医药产品;另一方面,该目录也应当参考国际经验,涵盖那些国际上容易引发公共健康危机的药品,特别是发展中国家和最不发达国家已实施强制许可的药品,我国亦可考虑列入。此外,对于指导目录内的药品,国家卫生健康委员会还可以考虑从单独实施招标采购、加快推动相关药品的使用等方面制定相应的指南。
3.建立国家知识产权局与国家药品监督管理局的协作程序
药品强制许可颁布后,国家药品监督管理局就成为了确保药品强制许可制度有效实施的核心部门。对此,国家药品监督管理局应当制定实施指南的相应附件,协同完成如下职责。第一,制定强制许可药品的优先审评审批规定。药品的优先审评审批规定应由国家药品监督管理局负责制定。具体而言,可以要求其在收到国家知识产权局的强制许可授予通报后,在保证相关仿制药的安全性、有效性和质量可控性的基础上,优先审评审批。同时,如果相关仿制药已先在欧美发达国家注册,可以考虑参考其数据,进一步加快审评审批进度。第二,确保药品上市许可期限与强制许可期限的一致。国家药品监督管理局颁发的药品批准文号,其有效期可以不按一般批准文号的期限执行,而应当与药品专利强制许可期限一致,以适应仿制药供应保障的要求。第三,加强强制许可药品的质量安全监管。国家药品监督管理局应对强制许可药品的制造、运输、销售、进口和出口等活动的全程监管,保障药品的质量安全。第四,鼓励高水平强制许可药品进行再注册或重新注册,保障其持续生产和供应。药品强制许可有效期届满前,如果被许可人能向药监部门提交其已获得专利许可的证明文件(再次获得的强制许可或获得的商业性专利许可)的,可按照药品注册相关规定优先审评审批并授予一般药品批准文号。若到期未能提供相关证明文件,药监部门应依法注销相关批准文号;但当相关药品专利到期后,该被许可人再申请注册的,药监部门应予受理并优先审评审批,尽快完成药品注册。
4.建立可强制许可药品的研发激励机制
鉴于涉及公共健康问题的专利药品多是治疗艾滋病、癌症等重大疾病领域品牌藥品,如果其技术水平仍然很高,那么国内仿制药企业这种“高水平仿制”本质上也应被认为是一种自主创新活动。在这方面,我国《科学技术进步法》第34条明确规定,“国家可以利用财政性资金设立基金,为企业自主创新与成果产业化贷款提供贴息、担保”,“政策性金融机构应当在其业务范围内对国家鼓励的企业自主创新项目给予重点支持”。有鉴于此,科技部可以考虑利用财政性资金设立基金,为我国仿制药企业的强制许可药品研发提供资金支持,推动相关药品的快速研发。在具体研发激励范围方面,除了艾滋病、恶性肿瘤、重大传染病等疾病的创新药应当受政府扶持,国内仿制药企业立足于国内地方性疾病、罕见病等药品的仿制研发活动也应当的得到重视。(谢莉琴、高星、胡红濮:《罕见病综合保障体系建设的国际经验与启示》,《社会保障研究》2018年第4期,第98—103页。)
药品专利强制许可制度是特殊情况下各国保障药品可及性的重要制度安排,其对于保障仿制药供应安全和应对公共健康危机的重要性已经得到了各国法治实践的充分检验。
实际上,在实行药品专利弱保护的印度等国,其主要是通过严格限制可专利性条件等方式控制药品专利的授权,(陈学宇:《交叉视角下药品标准必要专利常青的法律规制——以马来酸桂哌齐特注射液系列案件为例》,《法治论坛》2019年第1期。)再辅之以药品专利强制许可制度的实施,以求实现鼓励仿制药产业发展和保障药品可及性的目标。相比而言,我国的思路则不然。早在1992年《专利法》第一次修订时,我国就同步建立了药品专利制度和药品专利强制许可制度,试图通过鼓励药品创新和药品强制许可两套机制之间的协调,保障公众获得更多的药品。(程永顺、吴莉娟:《中国药品专利链接制度建立的探究》,《科技与法律》2018年第3期,第1—10页。)经过多年的发展,虽然我国医药产业的自主创新能力(特别是新药上市数量方面)已有了很大的提高,(姚雪芳、丁锦希、邵蓉、程璨:《中外创新药物研发能力比较分析——基于医药技术创新评价体系的实证研究》,《中国新药杂志》2010年第24期,第2231—2239页。)但是当前国内仍面临着仿制药供应结构性短缺、药品价格过高和公共健康问题频发的巨大压力。在此背景下,尽快根据我国公共健康状况和相关仿制药供应状况实施相应类型的药品专利强制许可,已是大势所趋。
现阶段,我国专利法上的药品专利强制许可制度的制度构造已经基本完成,药品专利强制许可一般制度与药品出口专利强制许可特殊制度的分类实施路径也初步建立。但是我国药品专利强制许可制度本身仍存在不少有待完善之处,不仅需要从分类实施的角度进行修正,还应当从药品专利强制许可实施全程管理的角度进行调整,方能保障其有效实施。当然,本文关于药品专利强制许可制度分类实施和全程管理的研究还比较粗浅,主要意在引起学界对这两个视角的关注;而对于不同类型药品专利强制许可的实施问题(例如未实施或未充分实施专利和为公共利益目的两种类型强制许可实施问题),笔者未来还将另行撰文进行专门探讨。
目前《专利法》第四次全面修订工作现已进入全国人民代表大会审议阶段,我国政府应当本着善意执行国际条约的原则,从我国公共健康状况和仿制药供应保障的现状出发,充分理解和运用TRIPS协议给予的弹性空间,借鉴其他国家的有益经验,进一步完善我国的药品专利强制许可制度,以期早日实现该制度的有效实施。
On Classified Implementation of Drug Patent CompulsoryLicense System in China
—— From the Perspective of Supply Security of Generic Drug
CHEN Xue-yu
[HJ]
Abstract: Drug patent compulsory license system, based on the balance between the patent rights and public interests, is an important system for many countries to reduce drug prices, improve drug accessibility and respond to public health crisis within the framework of the TRIPS Agreement. After three revisions, Chinas patent law has now established the basic framework of compulsory license system for drug patent, constructed two kinds of institutional arrangements: general system for compulsory licensing of drug patents and special system for compulsory licensing of drug export patents, designed five implementation paths, and initially established a supporting system for guaranteeing implementation. Institutional barriers are still the key factors hindering the implementation of compulsory license system for drug patents in china. The main problems are lack of operability on the general compulsory License system of drug patents, unreasonable design of content and function on the special system of compulsory license system of drug export patent, and the absence of supporting systems such as departmental collaboration and economic incentives. It is recommended that China should amend the relevant provisions of the Patent Law, improve the relevant contents of administrative regulations and departmental regulations, and improve the departmental cooperation mechanism based on the compulsory licensing system for drug patent as soon as posible.
Keywords: drug patent; compulsory license system; patent law; generic drug
【責任编辑 龚桂明 陈西玲】
猜你喜欢
中国知识产权(2019年4期)2019-05-08
法制与社会(2017年20期)2017-12-04
智富时代(2017年9期)2017-11-04
智富时代(2017年9期)2017-11-04
职工法律天地·下半月(2017年9期)2017-08-25
科技与创新(2017年11期)2017-07-01
法制博览(2017年4期)2017-05-20
学理论·下(2017年4期)2017-05-13
中国科技纵横(2017年5期)2017-05-12
进出口经理人(2016年15期)2017-05-10
