
Chk1及其抑制剂调控肿瘤作用机制研究
2019-11-15 08:36于祥菊宋碧莹刘滢吕冬霞
医学信息 2019年19期
关键词:抑制剂
于祥菊 宋碧莹 刘滢 吕冬霞
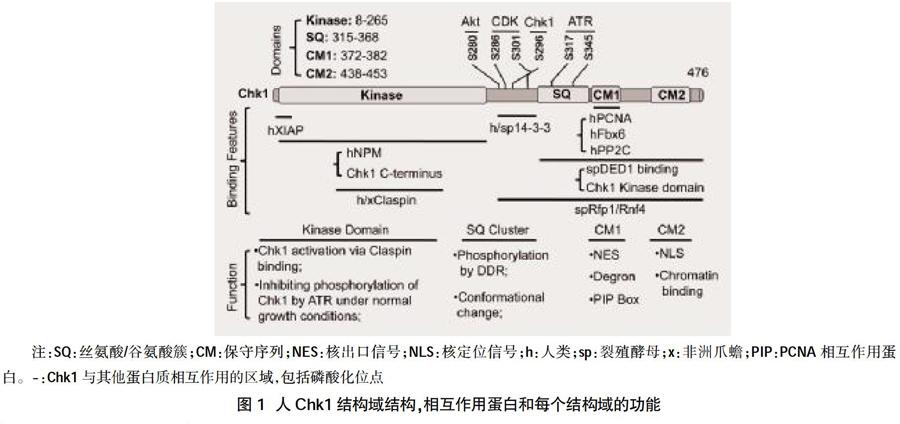
摘要:细胞周期检查点激酶1(Chk1)是由抑癌基因Chk1编码的丝氨酸/苏氨酸(Ser/Thr激酶),广泛存在于哺乳动物细胞内。近年来,多项国内外研究表明Chk1及其抑制剂能调控肿瘤的形成与发展。基于此,本文以Chk1及其抑制剂的概况和应用为基础,总结其多向调控肿瘤细胞的作用机制,旨在为Chk1及其抑制剂的临床应用和开发、相关药物的研发提供参考。
关键词:Chk1;抑制剂;肿瘤细胞;抑癌基因
中图分类号:R730.5 文献标识码:A DOI:10.3969/j.issn.1006-1959.2019.19.013
文章编号:1006-1959(2019)19-0039-05
Study on the Mechanism of Chk1 and Its Inhibitors Regulating Tumor
YU Xiang-ju,SONG Bi-ying,LIU Ying,LYU Dong-xia
(Jiamusi University,Jiamusi 154007,Heilongjiang,China)
Abstract:Cell cycle checkpoint kinase 1 (Chk1) is a serine/threonine (Ser/Thr kinase) encoded by the tumor suppressor gene Chk1 and is widely found in mammalian cells. In recent years, a number of domestic and foreign studies have shown that Chk1 and its inhibitors can regulate the formation and development of tumors. Based on this, based on the profile and application of Chk1 and its inhibitors, this paper summarizes the mechanism of multi-directional regulation of tumor cells, aiming to provide reference for the clinical application and development of Chk1 and its inhibitors, and the development of related drugs.
Key words:Chk1;Inhibitor;Tumor cells;Tumor suppressor gene
肿瘤是由遗传物質改变导致细胞过度增殖而形成的赘生物。传统的治疗方法多首选外科治疗,加以辅助放射治疗或化学药物治疗。现今,肿瘤治疗已逐渐进入分子领域。靶向肿瘤增殖、扩散位点的抑制剂也已广泛应用,明显提高了患者的生存周期和质量。已有研究证实,DNA损伤应答信号网络(DNA damage response,DDR)是肿瘤形成过程中的主要调节机制[1]。而在DDR中,Chk1是除p53外诱导DNA损伤最常见的核心蛋白[2]。到目前为止,关于Chk1及其抑制剂调控肿瘤的作用机制的研究引起广泛关注。本文主要总结Chk1抑制剂调控肿瘤的作用机制,以期扩展Chk1的相关药物研发。
1 Chk1概述
Chk1蛋白属于Ser/Thr激酶家族,分子量约为54 kD。由人11号常染色体长臂2区4带2亚带上的基因编码。cDNA全长约1891 bp,内含16个外显子。Chk1具有高度保守的、能催化底物的 N端激酶结构域和200个残基构成的抑制性的C端调节结构域(图1)。其同源物中间区还接有可变连接域以及作为ATM和ATR激酶的催化底物的SQ/TQ调节域[1,2]。Chk1通过磷酸化下游分子触发多效应细胞反应,包括转录调节、细胞周期停滞或延迟、DNA损伤修复甚至诱导细胞死亡。
1.1 Chk1与细胞周期 细胞周期是高度保守、有序进行的细胞增殖过程。人的细胞周期由细胞分裂间期中的G1、S和G2期以及分裂期M期构成。最早证实Chk1能控制G2/M期转换,是裂殖酵母中CDK1/cdc2的突变体基因[3]。Chk1被阻断时,紫外线诱导的酵母细胞DNA损伤更易发生,DNA复制停滞在G2期[4]。随后在果蝇、人和小鼠中先后发现了Chk1的同源物,其能调节细胞分裂的保真度、有丝分裂以及胚胎发育过程。在非洲爪蟾胚胎中,Chk1以母体/合子基因产物方式瞬时激活,通过靶向cdc25A降解以及SCFβ-TRCP E3泛素连接酶磷酸化降解调节MBT互质因子Drf1的水平,进而参与中胚轴转变(MBT)以来延长细胞周期[5,6]。Smith J等[7]通过培育Chk1等位基因纯合子的C57BL/6小鼠,发现黑色素细胞在胚胎发育时形成,但在Tyr:Cre表达开始的几天后逐渐丢失,表明重组后的Chk1蛋白异常消耗导致细胞死亡。Lunardi A等[8]证实ETS转录因子家族能直接抑制前列腺癌中Chk 1的转录促进肿瘤发生。研究表明ETS转化潜力是通过抑制Chk1的表达,进而促进未修复DNA损伤过程激发的基因组不稳定性。进一步研究显示,磷酸化的Chk1与14-3-3蛋白结合募集到E2F7/8阻遏产物的启动子区域干扰转录过程,促进ETS家族中非典型E2F表达,进而永久性阻滞S期抑制肿瘤形成[9]。
1.2 Chk1与DDR DDR是细胞内和响应DNA损伤的重要调节信号网络系统,可协调、检测和修复DNA损伤过程。损伤后的DNA一部分能间接通过DDR响应周期活化,另一部分直接修复损伤。若双链DNA断裂,ATM/ Chk2蛋白激酶通路活化进行修复[10]。而单链DNA(ssDNA)的损伤能通过激活ATR/Chk1途径,磷酸化Ser-317号和Ser-345号位点执行DNA损伤检测途径[11]。由于Chk1是ATR依赖的DDR反应的关键组分,通过组织复制叉塌陷和激活DDR保护细胞免受复制应激的侵害,被认为是肿瘤抑制因子。Lam MH等[12]构建了Chk条件表达小鼠,发现在单倍体Chk1+/-上皮细胞中,细胞周期异常增殖,S期细胞更早进入有丝分裂过程,表现出恶性肿瘤的征兆。但目前尚未在人类肿瘤中发现Chk1完全丧失或突变的纯合子,可见作为肿瘤抑制因子Chk1的抑制作用较弱。
2 Chk1 抑制剂
作为DDR的核心,关于靶标Chk1的化疗药物研究不断深入。起初认为Chk1被抑制时,癌细胞特别是p53缺失的肿瘤细胞会丧失响应和修复DNA损伤的能力,增强放化疗法对细胞的杀伤作用。因此,早期Chk1抑制剂集中在与放疗或化疗结合参与癌症治疗过程。1995年,第一个开发出的Chk1抑制劑——7-羟基星形孢菌素(UCN-01)进入Ⅰ期临床试验。目前已证实其能直接作用于特异性磷酸酶CDC25C和AKT上游的PDK1从而停滞细胞周期[13]。此外,Lien WC等[14]发现UCN-01能诱导人骨肉瘤U2~OS细胞周期停滞、损伤DNA,并激活非经典下游效应物ERK抑制肿瘤细胞增殖。随后,不同公司开发了数十种小分子Chk1抑制剂,对Chk1具有更高的效力和更高的特异性。
AZD7762是一种有效的Chk1/2的双重抑制剂,属于ATP竞争性抑制剂。在人乳腺癌4T1.2细胞中,AZD7762作用后Caspase-3,LC3A/BⅡ,p-eIF2的表达升高而p-Chk1和pChk2的表达降低。证明其能通过诱导自噬和凋亡协同促进肿瘤细胞增殖[15]。除了抑制作用,Park YH等[16]研究发现AZD7762能抑制肥大细胞中的Syk及其下游信号蛋白如丝裂原活化蛋白激酶Erk1/2,磷脂酶(PL),Cy1的活化发挥体外抗过敏作用。MK-8116也叫SCH900776,其属于高选择性的Chk1抑制剂,可削弱DNA修复能力主要用于乳腺癌和白血病的治疗。与第一代Chk1抑制剂UCN-01相比,其毒性更低,能诱导中心体突变,同时显著停滞G2/M检查点,延长有丝分裂期并增加异常分裂细胞数[17]。LY2603618是一种有效的选择性Chk1蛋白激酶小分子抑制剂,可用于人非小细胞肺癌、胰腺癌细胞。Laquente B等[18]的65例LY2603618处理的胰腺癌患者的Ⅱ期临床研究证明,使用LY2603618后胰腺癌患者的总生存期效果不如吉西他滨,若联合应用效果可以提高30%。
除了上述常见的Chk1抑制剂,新型的Chk1抑制剂仍然处于临床开发阶段,目前主要为ATP竞争抑制剂,包括V158411,CCT245737,GDC-0575等。Wayne J等[19]在人类癌症模型中发现了V158411作用促进肿瘤细胞作用机制。从分子水平上看,V158411诱导DNA合成过程中γH2AX阳性细胞核呈现时间和浓度依赖性增加。并通过γH2AX表达诱导ATR/ATM/DNA-PKcs DNA损伤应答途径的激活。CCT245737是临床开发的第一个口服活性的候选CHK1抑制剂,现已进入Ⅰ期临床实验,其IC50为1.4 nM,对CHK2和CDK1的选择性>1000倍。CCT245737能抑制多种人肿瘤细胞系中基因毒性诱导的CHK1活性(pS296CHK1)和细胞周期停滞(pY15CDK1)的表达,导致DNA损伤和细胞凋亡增加[20]。GDC-0575为近年来较新型的Chk1抑制剂。Laroche A等[21]发现GDC-0575消除了DNA损伤诱导的S期和G2/M检查点,加剧了DNA双链断裂并诱导软组织肉瘤STS细胞凋亡。此外,若其与吉西他滨在TP53缺陷的肉瘤模型中联合应用可出现协同或累加效应。Tullio AD等[22]在白血病(AML)中的IC50为1.2 nM,其能阻断阿糖胞苷(AraC)激活磷酸化的CDK2诱导的Chk1的表达,协同治疗白血病。
3 Chk1抑制抗肿瘤作用机制
众所周知,多数传统抗癌药物的作用靶标主要为DNA,可通过直接损害DNA或阻碍其前体的合成来消灭肿瘤细胞。在DNA受损过程中,DDR通路活化修补损伤的DNA促使肿瘤细胞继续增殖。因此Chk1抑制剂恰好弥补传统药物的缺陷,成为抗肿瘤药物研究热点。目前已开发的Chk1抑制剂既可以单独应用,也能协同联合放疗或化疗药物参与临床抗肿瘤研究,其相关机制也不断完善。
3.1单一作用 起初Chk1抑制剂主要用于治疗肺癌、黑色素瘤、膀胱癌等恶性程度较高的肿瘤。其单一作用机制表现为阻滞细胞周期、损伤DNA等。Wayne J等[23]设计并开发了新型口服Chk1抑制剂VER-250840,体外研究实验显示该抑制剂24 h内pChk1抑制率超过90%,并损伤基因组DNA使黑色素瘤A2058细胞出现不可逆性细胞周期停滞。目前,已进入Ⅱ期实验的Chk1抑制剂prexasertib能通过增加复制应激促进DNA损伤过程,诱导非小细胞肺癌分裂过程提前终止。但却不可避免的出现化疗药物耐药性。Deraska P等[24]研究发现如果敲除FAM122A蛋白,prexasertib的抗性解除,PP2A蛋白表达增加发挥抗癌活性。此外, CCT244747抑制剂能增加PARP裂解,通过Caspase-3活化诱导膀胱癌T24细胞系和人舌鳞癌Cal27细胞系凋亡[25]。Zhou ZR等[26]首次提出MK-8776作用于人三阴性乳腺癌(TNBC)MDA-MB-231,BT-549和CAL-51细胞后,与单独照射相比,低剂量的抑制剂能增加放疗致敏性并增加3种TNBC细胞中诱导的γH2AX的表达,提示对DNA损伤增强抑制细胞增殖。随着时间的延长透射电镜检测发现MK-8776作用后能增强自噬体的数量促进LC3-Ⅰ转化为LC3-Ⅱ。在UCN-01处理人结肠癌HCT116等肿瘤细胞后,通过Akt-FoxO3a途径激活Puma诱导线粒体凋亡途径。同时期也可触发Caspase-9直接激活Caspase-3,进而通过正反馈环路切割XIAP 发挥抗肿瘤作用[27]。Wang FZ等[28]研究证实LY2603618后DDR明显诱导表现为ATM,Chk2,p53和组蛋白H2AX的磷酸化,同时能抑制S296 Chk1和增加DNA损伤介导的S345 Chk1的表达。
3.2联合应用 除了与放疗合用,其他许多化疗剂如抗代谢物质或铂类相关药物等也参与激活复制应激,引起激活CHK1信号传导的DNA损伤。因此,CHK1抑制剂已被评估为电离辐射(IR)的敏感剂和多种人类癌症模型中的细胞毒性药物。相关课题组通过CHK1抑制剂在体外和体内的生物学评价揭示了它们与放化疗等组合的活性以及相关作用机制。
3.2.1增加放疗敏感性 放射疗法(RT)是通过IR造成多种形式的DNA损伤从而杀死恶性肿瘤的常规治疗方式。但经过多次放射后,肿瘤细胞能形成适应性反应进而产生RT抵抗。如今已在多种肿瘤模型中证实Chk1抑制剂能增强放疗的敏感性。Zhang Y等[29]报道癌基因c-Myc/CDC25A/c-Src/H-ras/E2F1以及DDR蛋白ATR/CHK1/BRCA1/CtIP在放射抗性乳腺癌细胞(RBCC)中表达上升。Chk1抑制剂AZD7762治疗后,DNA消耗增加,复制叉进展速度显著降低,同时中断复制叉稳定性与同源重组过程,增加放射敏感性。此外,不同的Chk1抑制剂暴露在IR后对细胞毒性的作用机制不同。Suzuki M等[30]發现新型选择性抑制剂MK-8776不同于传统抑制剂UCN-01影响DNA双链的作用机制,而是通过激活纺锤体组装检查点增加有丝分裂缺陷。
3.2.2协同其他化疗药物 相较于Chk1抑制剂的单一作用,关于Chk1抑制剂与其他分子靶向剂协同抗肿瘤的研究更为深入。目前已发现Chk1抑制剂可与抗代谢药物、铂类化疗药物以及其他靶向抑制剂如PAPP抑制剂等结合。并进一步研究揭示相关组合的效应机制,弥补单一抗癌药物缺陷。吉西他滨隶属嘧啶类抗肿瘤药,主要通过阻断核酸的合成(S期)治疗肺癌、乳腺癌等疾病。Barnard D等[31]发现单独使用吉西他滨诱导肿瘤细胞Chk1磷酸化,副作用较大。当两者联合治疗后,LY2603618能加强阻断S期检查点联合诱导肠癌、肺癌和胰腺癌异种移植模型肿瘤的生长。Vincelette ND等[32]报道在急性髓系白血病(AML)U937细胞中,Chk1抑制剂MK-8876能降低CPX-351诱导的Chk1 Ser-296号、KAP 1 Ser-473号以及CDC25A磷酸化水平,自动降解复制检查点蛋白,并诱导细胞凋亡进而增强对原发性AML的抗增殖作用。此外,SB218078抑制剂与奎纳克林不仅能阻断G2/M期促进有丝分裂过程中断,还能通过破坏乳腺癌细胞BER通路进而诱导细胞凋亡[33]。
此外,将Chk1抑制剂与靶向DDR位点药物结合的研究引起广泛关注。主要包括WEE1抑制剂、PAPP抑制剂等方面。Hauge S等[34]研究发现单独使用WEE1抑制剂MK1775能使S期CDK活性增加,联合给与Chk1抑制剂负向调控复制因子Treslin表达,抑制CDC45负荷,从而限制S期DNA损伤程度,起到协同抗癌的作用。Booth L等[35]报道显示Chk1抑制剂SRA737与PARP抑制剂niraparib联合后自噬相关底物p62和LAMP62水平降低,同时ATG5、p-ATG13以及Beclin1表达增加。两者协同经mTOR-AMPK-ULK途径发挥杀伤性自噬作用,或经Bcl-XL内在凋亡通路抑制肿瘤增长。另有研究显示,LY606368与PARP1抑制剂BMN673协同促进胃癌经Caspase经典途径凋亡,并抑制同源重组修复加速细胞死亡[36]。
4总结
开发越来越有效和特异性的Chk1抑制剂在临床实验中逐渐受到重视。Chk1是癌症对常规治疗的选择性致敏的良好靶标。本文总结了Chk1的功能效应及其在肿瘤中的作用机制,在此基础上,进一步分析Chk1抑制剂的进展与肿瘤关系的作用机制。总之,了解Chk1及其抑制剂与肿瘤发生发展的作用机制有助于构建新一代Chk1抑制剂弥补现今肿瘤治疗的缺陷。最后,想要产生新型的Chk1抑制剂,还要在临床试验中获得成功测试,不断寻找优化的方案以提高其功效,并鉴定适合这些药剂的生物标志物等,从而在正在进行的以及未来的临床试验中完全阐明Chk1抑制剂在癌症治疗中的作用。
参考文献:
[1]Zhang Y,Hunter T.Roles of Chk1 in cell biology and cancer therapy[J].Int J Cancer,2014,134(5):1013-1023.
[2]Walker M,Black EJ,Oehler V,et al.Chk1 C-terminal regulatory phosphorylation mediates checkpoint activation by de-repression of Chk1 catalytic activity[J].Oncogene,2009,28(24):2314-2323.
[3]Walworth N,Davey S,Beach D.Fission yeast chkl protein kinase links the rad checkpoint pathway to cdc2[J].Nature,1993,363(6427):368-371.
[4]Furuya K.DNA checkpoints in fission yeast[J].Journal of Cell Science,2003,116(19):3847-3848.
[5]Shimuta K,Nakajo N,Uto K,et al.Chk1 is activated transiently and targets Cdc25A for degradation at the Xenopus midblastula transition[J].The EMBO J,2001,21(14):3694-3703.
[6]Collart C,Smith JC,Zegerman P.Chk1 Inhibition of the Replication Factor Drf1 Guarantees Cell-Cycle Elongation at the Xenopus laevis Mid-blastula Transition[J].Developmental Cell,2017,42(1):82-96.
[7]Smith J,Larue L,Gillespie DA.Chk1 is essential for the development of murine epidermal melanocytes[J].Pigment Cell Melanoma Res,2013,26(4):580-585.
[8]Lunardi A,Varmeh S,Chen M,et al.Suppression of CHK1 by ETS Family Members Promotes DNA Damage Response Bypass and Tumorigenesis[J].Cancer Discovery,2015,5(5):550-563.
[9]Yuan R,Vos HR,Van RE,et al.Chk1 and 14-3-3 proteins inhibit atypical E2Fs to prevent a permanent cell cycle arrest[J].Embo Journal,2018,37(5):1-17.
[10]O'Connor MJ.Targeting the DNA Damage Response in Cancer[J].Molecular Cell,2015,60(4):547-560.
[11]Liu Y,Vidanes G,Lin YC,et al.Characterization of a Saccharomyces cerevisiae homologue of Schizosaccharomyces pombe Chk1 involved in DNA-damage-induced M-phase arrest[J].Mol Gen Genet,2000,262(6):1132-1146.
[12]Lam MH,Liu Q,Elledge SJ,et al.Chk1 is haploinsufficient for multiple functions critical to tumor suppression[J].Cancer Cell,2004,6(1):45-59.
[13]Signore M,Buccarelli M,Pilozzi E,et al.UCN-01 enhances cytotoxicity of irinotecan in colorectal cancer stem-like cells by impairing DNA damage response[J].Oncotarget,2016,7(28):44113-44128.
[14]Lien WC,Chen TY,Sheu SY,et al.7-hydroxy-staurosporine,UCN-01,induces DNA damage response, and autophagy in human osteosarcoma U2-OS cells[J].Journal of Cellular Biochemistry,2018,119(6):4729-4741
[15]Luqi W,Yue W,Andy C,et al.Effects of a checkpoint kinase inhibitor,AZD7762,on tumor suppression and bone remodeling[J].International Journal of Oncology,2018,53(3):1001-1012.
[16]Park YH,Kim DK,Kim HW,et al.Repositioning of anti-cancer drug candidate,AZD7762,to an anti-allergic drug suppressing IgE-mediated mast cells and allergic responses via the inhibition of Lyn and Fyn[J].Biochem Pharmac,2018,154(1):270-277.
[17]Suzuki M,Yamamori T,Bo T,et al.MK-8776,a novel Chk1 inhibitor,exhibits an improved radiosensitizing effect compared to UCN-01 by exacerbating radiation-induced aberrant mitosis[J].Translational Oncology,2017,10(4):491-500.
[18]Laquente B,Lopez-Martin J,Richards D,et al.A phase Ⅱ study to evaluate LY2603618 in combination with gemcitabine in pancreatic cancer patients[J].BMC Cancer,2017,17(1):137-146.
[19]Wayne J,Brooks T,Massey AJ.Inhibition of Chk1 with the small molecule inhibitor V158411 induces DNA damage and cell death in an unperturbed S-phase[J].Oncotarget,2016,7(51):85033-85048.
[20]Walton MI,Eve PD,Hayes A,et al.The clinical development candidate CCT245737 is an orally active CHK1 inhibitor with preclinical activity in RAS mutant NSCLC and Eμ-MYC driven B-cell lymphoma[J].Oncotarget,2016,7(3):2329-2342.
[21]Larocheclary A,Lucchesi C,Rey C,et al.CHK1 Inhibition in Soft-Tissue Sarcomas:Biological and Clinical Implications[J].Annals of Oncology Official Journal of the European Society for Medical Oncology,2018,29(4):1023-1029.
[22]Tullio AD,Rouault-Pierre K,Abarrategi A,et al.The combination of CHK1 inhibitor with G-CSF overrides cytarabine resistance in human acute myeloid leukemia[J].Nature Communications,2017,8(1):1679.
[23]Wayne J,Stokes S,Foloppe N,et al.Identification and preclinical characterisation of VER-250840,a potent,selective Chk1 inhibitor with in vivo oral single-agent antitumor activity [J].Molecular Targets and Cancer Therapeutics,2018,17(1):Abstract nr B163.
[24]Deraska P,Reavis H,Labe S,et al.Whole-genome CRISPR/Cas9 screen of the CHK1 inhibitor prexasertib implicates FAM122A loss as a potential resistance mechanism[J].AACR Cancer Res,2018,78(13):4287.
[25]Patel R,Barker HE,Kyula J,et al.An orally bioavailable Chk1 inhibitor, CCT244747, sensitizes bladder and head and neck cancer cell lines to radiation[J].Radiotherapy and Oncology,2017,122(3):470-475.
[26]Zhou ZR,Yang ZZ,Wang SJ,et al.The Chk1 inhibitor MK-8776 increases the radiosensitivity of human triple-negative breast cancer by inhibiting autophagy[J].Acta Pharmacol Sin,2017,38(4):513-523.
[27]Nie C,Luo Y,Zhao X,et al.Caspase-9 mediates Puma activation in UCN-01-induced apoptosis[J].Cell Death and Disease,2014,5(10):e1495.
[28]Wang FZ,Fei HR,Cui YJ,et al.The checkpoint 1 kinase inhibitor LY2603618 induces cell cycle arrest,DNA damage response and autophagy in cancer cells[J].Apoptosis,2014,19(9):1389-1398.
[29]Zhang Y,Lai J,Du Z,et al.Targeting radioresistant breast cancer cells by single agent CHK1 inhibitor via enhancing replication stress[J].Oncotarget,2016,7(23):34688-34702.
[30]Suzuki M,Yamamori T,Bo T,et al.MK-8776,a novel Chk1 inhibitor,exhibits an improved radiosensitizing effect compared to UCN-01 by exacerbating radiation-induced aberrant mitosis[J].Translational Oncology,2017,10(4):491-500.
[31]Barnard D,Diaz HB,Burke T,et al.LY2603618,a selective CHK1 inhibitor,enhances the anti-tumor effect of gemcitabine in xenograft tumor models[J].Investigational New Drugs,2016,34(1):49-60.
[32]Vincelette ND,Ding H,Huehls AM,et al.Effect of CHK1 Inhibition on CPX-351 Cytotoxicity in vitro and ex vivo[J].Scientific Reports,2019,9(1):3617.
[33]Preet R,Siddharth S,Satapathy SR,et al.Chk1 inhibitor synergizes quinacrine mediated apoptosis in breast cancer cells by compromising the base excision repair cascade[J].Biochemical Pharmacology,2016,105(1):23-33.
[34]Hauge S,Naucke C,Hasvold G,et al.Combined inhibition of Wee1 and Chk1 gives synergistic DNA damage+in S-phase due to distinct regulation of CDK activity and CDC45 loading[J].Oncotarget,2017,8(7):10966-10979.
[35]Booth L,Roberts J,Poklepovic A,et al.The Chk1 inhibitor,SRA737,synergizes with niraparib to kill cancer cells via multiple cell death pathways[A]//Proceedings of the American Association for Cancer Research Annual Meeting[C].2018.
[36]Yin Y,Shen Q,Zhang P,et al.Chk1 inhibition potentiates the therapeutic efficacy of PARP inhibitor BMN673 in gastric cancer[J].American Journal of Cancer Research,2017,7(3):473.
猜你喜欢
天然产物研究与开发(2018年7期)2018-08-21
上海农业学报(2017年3期)2017-04-10
西南国防医药(2016年6期)2016-12-01
数学年刊A辑(中文版)(2016年2期)2016-10-30
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
中国民族医药杂志(2016年2期)2016-05-14
国外医药(抗生素分册)(2015年3期)2015-07-12
中国当代医药(2015年20期)2015-03-01
中国当代医药(2015年16期)2015-03-01
