
神经疾病诊断与研究中的神经递质检测技术
2019-11-12 06:29都展宏鲁艺王立平
分析化学 2019年10期
都展宏 鲁艺 王立平

摘 要 神经递质的在体检测不仅可能作为帕金森氏症、阿尔兹海默症、抑郁症等多种神经疾病的生物标记物,同时,这类传感器也可作为研究奖赏、成瘾、厌恶等多种行为产生机制的重要工具。近年来,多种检测神经递质的分析技术已得到了长足的发展,但目前应用于疾病研究和在体检测的传感技术与材料尚面临一些困难,只有较少的应用实例。本文主要介绍了癫痫、帕金森氏症、脑卒中、阿尔兹海默症、精神分裂等疾病中神经递质发挥的重要作用及其检测的科学意义,探讨了在体传感器技术的进展,以及可能用于解析疾病神经环路机制的相关技术和器件,重点探讨了多种纳米材料、导电聚合物材料和生物分子作为在体传感器组成成分的应用技术,以及未来这些新技术、新材料面临的机遇与挑战。
关键词 神经递质; 生物传感器; 纳米材料; 成像技术; 疾病诊断; 评述
1 引 言
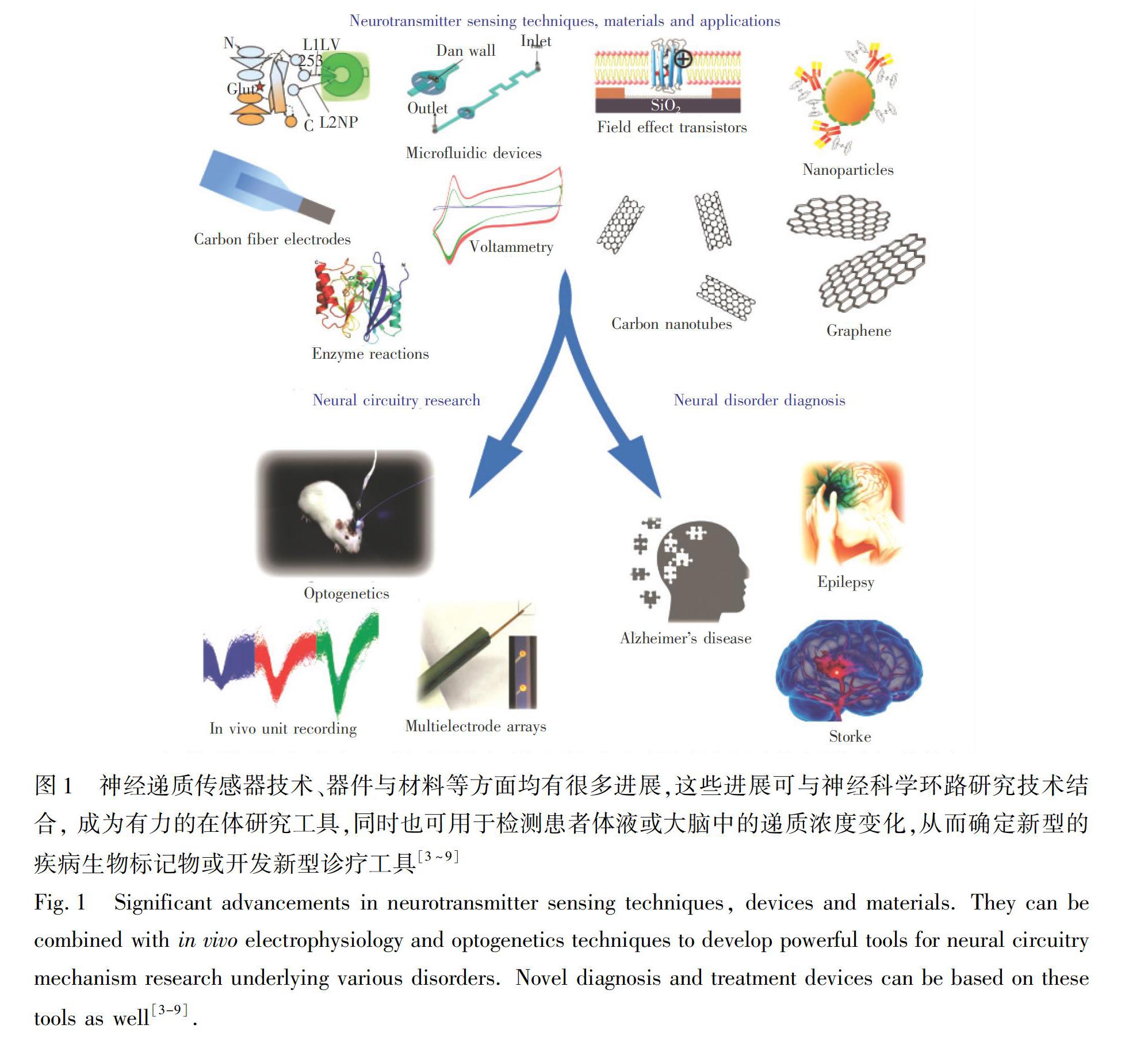
神经递质是体内负责在神经元与其它细胞类型间传递或调控特定信号的化学分子。常见的神经递质分子包括谷氨酸、γ 氨基丁酸、多巴胺、乙酰胆碱、肾上腺素、血清素、去甲肾上腺素和一氧化氮等[1]。神经递质分子在行为与认知发生等脑功能中都发挥着重要作用; 同时,它们在调控心率、学习记忆、睡眠、觉醒、意识、情绪和食欲等方面具有重要意义。以谷氨酸为例,它是大脑最重要的兴奋性神经递质,在学习记忆、大脑发育、情绪情感调节等过程中都发挥了重要作用。谷氨酸能神经元的异常也与多种疾病相关,如神经退行性疾病和精神类疾病等[2]。本文重点介绍了一些重要神经递质分子在多种神经系统疾病中的作用机制,如图1所示[3~9]。本文还介绍了神经递质检测技术和器件、目前用于神经系统疾病相关的环路研究机制、为满足疾病诊疗需求开发的相关技术,以及为递质传感器界面开发的多种类别的电极材料。最后,简要介绍了这些递质传感技术在疾病与神经科学应用中面临的机遇与挑战。
2 神经递质在脑功能与神经疾病中的作用
各种神经递质分子在神经系统的活动中各自发挥着不同的作用,并且很多神经疾病都以一种或数种神经递质系统的突触传递发生改变为特征。神经递质检测技术既可帮助研究者更好地理解这种递质在生理和病理方面的作用,又可有效地验证针对特定神经递质系统的治疗方法与某些特定的疗效或行为改变的相关性。 以下对5种高发的神经疾病及其密切相关的神经递质系统功能异常做简要介绍,这些病理神经递质系统有可能成为神经递质传感器重要的在体应用研究方向。
2.1 癫痫
大脑中最普遍的兴奋性神经递质谷氨酸和最普遍的抑制性神经递质γ 氨基丁酸在多数脑区和核团中都普遍存在,也介导了多数长程神经投射的功能,而谷氨酸与γ 氨基丁酸对神经功能的作用最直接、最显著,其中研究最多的疾病就是癫痫。在患者大脑中,一些细胞集群活动的增强和同步可能导致癫痫发作,并且,有些细胞亚群可能会产生内源性的爆发性发放状态,这些机制的产生可能与γ 氨基丁酸、谷氨酸作用机制密切相关,特别是NMDA受体介导的兴奋性谷氨酸电流[10,11]。这些兴奋性和抑制性神经递质系统在癫痫疾病发作中的异常,特别是其与癫痫起始、持续放电和传播之间的关系已有大量的经典研究支持[12~14]。然而,这种过度的兴奋性发放究竟是由于γ 氨基丁酸能神经元死亡而导致的抑制性活動减弱,还是由于γ 氨基丁酸本身释放减少或者受体减少导致突触传递功能减弱,目前仍然不清楚。
在脑内直接给予谷氨酸或者其受体激动剂(如N 甲基 D 天冬氨酸(N methyl D aspartate,NMDA))即可直接诱发癫痫。并且,大多数神经元的谷氨酸浓度可达到10 mmol/L,远高于其它兴奋性递质的浓度。一旦神经细胞受损,胞内谷氨酸就会泄漏出来,而胞外普遍存在谷氨酸受体。另外,一旦神经组织的结构产生病变,如脑中的瘢痕组织、肿瘤的发生和生长,以及其它产生胶质细胞病变的情况,都很可能损伤胞外的高效率谷氨酸转运重吸收系统[15],这些系统能稳定地将胞外的谷氨酸浓度维持在5 mmol/L以下,最多不超过10 mmol/L。
相关研究表明,在癫痫灶点位置γ 氨基丁酸可使神经元或神经末梢有一定的损伤[16,17],但也有人体组织研究表明,患者海马体内γ 氨基丁酸神经元的群体未见变化[18]。动物模型中,点燃癫痫模型的大鼠杏仁核内γ 氨基丁酸能神经元或神经末梢显著下降(约30%~50%),杏仁核内γ 氨基丁酸的浓度也观测到类似比例的下降[19,20]。作为颞叶癫痫密切相关的脑区,海马与杏仁核的递质系统病变对于癫痫研究有重要的指示作用。有研究表明,在人体颞叶癫痫灶点位置手术切除的组织中,用神经化学检测的方式可测得γ 氨基丁酸能突触传递明显下降[21,22],然而现有研究未能证实该结果[18]。有研究证明,点燃癫痫模型大鼠的杏仁核胞外微透析实验测得的γ 氨基丁酸浓度显著下降了60%~70% [23],一定程度上反映了癫痫动物中枢神经系统中γ 氨基丁酸的合成明显降低。针对兴奋/抑制递质系统失衡的特点,长期的经验和研究表明,采用可影响胞外γ 氨基丁酸代谢的药物增加中枢神经系统中的γ 氨基丁酸浓度, 可作为抑制癫痫的手段,但通过谷氨酸脱羧酶(Glutamic acid decarboxylase, GAD)抑制剂降低γ 氨基丁酸浓度, 则会导致癫痫发生。
2.2 帕金森氏症
帕金森氏症的病理特征中,最显著的标志是在所有向端脑脑区投射的多巴胺能神经元突触前化学递质的减少[24~26]。这种多巴胺能投射减弱的最主要原因是黑质及其相关的黑质旁核中的多巴胺能神经元胞体减少。研究表明,密集投射到纹状体、伏隔核、嗅觉脑区和边缘皮层区域的多巴胺能神经纤维主要源自黑质的致密区[27,28]。一旦以药物抑制或物理损伤等方式破坏黑质投射到基底神经节的多巴胺能神经元,基底神经节的正常功能就会受到严重影响。无论在动物模型, 还是人体中,这种纹状体中多巴胺能投射的减少都介导了帕金森氏症中的主要症状(运动迟缓、肌强直、静止性震颤)。在帕金森氏症患者脑内的黑质、尾状核、壳核、苍白球等脑区中,无论是多巴胺浓度, 还是其代谢产物(高香草酸)的浓度,都明显减少[29~31]。在原发性帕金森氏症中,整体多巴胺损伤程度略轻,并且壳核受损比尾状核严重[29]; 而在脑炎性帕金森病症中,多巴胺能投射均有相对均匀的严重损伤。这种差异的主要原因为脑炎型帕金森氏症中,黑质多巴胺神经元受到普遍的严重损伤,而原发性帕金森氏症中的黑质多巴胺神经元损伤相对较轻,且主要影响特定区域的多巴胺能神经元胞体,如尾侧黑质中的多巴胺能神经元[29],而这些神经元主要投射到壳核中。因此,在多巴胺能投射丰富的脑区测定多巴胺浓度的空间分布对研究各种症状的环路机制和开发针对性诊疗工具都具有重要的作用。
2.3 脑卒中
脑卒中是另一大类发作率非常高的神经系统疾病,谷氨酸兴奋性毒性导致的皮层损伤是卒中最严重的后果之一[32]。 尽管卒中方面的基础研究已取得了很多进展,临床上针对这种严重病情所做的各种治疗和预防尝试都还未能达到很好的效果。在脑缺血发生后,神经元释放的大量谷氨酸会激活多种突触前和突触后的谷氨酸受体,从而导致胞内Ca2+浓度上升,进而导致氧自由基产生,线粒体功能异常,蛋白酶、磷酸酯酶、限制性内切酶等多种酶的激活,从而造成细胞死亡。同时,脑缺血发生后,兴奋性氨基酸转运体(Excitatory amino acid transporter 1,EAAT1)和EAAT2均有明显损失,造成了胞外谷氨酸浓度升高,最终导致神经元死亡[33~35]。另一种重要的细胞星形胶质细胞对长时间的乳酸中毒非常敏感[36],直接导致它们在酸性环境无法维持三磷酸腺苷(Adenosine triphosphate,ATP)产生[37]。ATP浓度下降进一步导致星形胶质细胞的跨膜离子浓度梯度无法维持,钠钾离子ATP酶的活性被抑制,进而导致Na+内流,胞内Na+浓度升高,星形胶质细胞胞内的谷氨酸由于其转运受到Na+浓度的影响而被转移到胞外[38],相当于谷氨酸转运体的功能被逆转[39 41]。这一过程与神经细胞的谷氨酸系统异常联合将导致胞外谷氨酸浓度进一步升高。因此,在相对温和的脑缺血状态下,星形胶质细胞通过谷氨酸转运体将一些谷氨酸转移到胞内,实现神经保护,但在严重缺血状态下, 星形胶质细胞会起反作用,进一步加剧毒性[42]。在脑缺血的过程中,大脑白质中的细胞会受到严重影响,少突胶质细胞和它们产生的髓鞘会受到兴奋性毒性的严重影响[43~47],而白质中的轴突则会由于胞内Ca2+浓度剧烈升高导致的毒性而严重受损[48~50]。
2.4 阿尔兹海默症
关于阿尔兹海默症的理论已有多方面的研究,其中一种与神经递质系统功能异常密切相关的神經毒性理论认为,当局部谷氨酸浓度过高时,会对脑组织产生毒性[51]。一种假说认为,谷氨酸兴奋性毒性会激活代谢型谷氨酸受体及下游的一系列分子通路,导致阿尔兹海默症的发生。同时,这些通路也被认为是可能的药物治疗靶点,因此,针对代谢型谷氨酸受体的调控通路,研究者也在开发多种阿尔兹海默症的潜在治疗药物[52]。这些受体下游有大量复杂的G蛋白偶联受体相关通路,因此可能的调控干预方式也存在各种差别。另一种对阿尔兹海默症十分重要的神经递质分子乙酰胆碱不仅在大脑中发挥重要的作用,也在脊髓和其它组织中调控多种重要功能。 分泌乙酰胆碱的神经元被称为胆碱能受体, 而乙酰胆碱在认知和运动等多个功能的执行中都有不可替代的作用。近年,针对谷氨酸和乙酰胆碱等递质开发了一系列基于酶反应的体内及体外生物传感器系统[53]。而主流的研究认为,脑内乙酰胆碱含量增多与阿尔兹海默症的症状改善密切相关,因而多种乙酰胆碱和谷氨酸传感器均有可能在阿尔兹海默症相关研究中发挥重要作用。
2.5 精神分裂症
一种重要的精神分裂症成因假说是谷氨酸能神经元的功能弱化导致该病发生[54]。在患者脑脊液中测得的谷氨酸浓度降低及谷氨酸拮抗剂可导致部分症状产生的现象均可在一定程度证明该假说。特别是盐酸苯环己哌啶的使用可诱导一种类似于精神分裂症的疾病状态, 使得该假说被相对广泛地接受[55,56]。另一方面,苯环己哌啶及类似物(如盐酸氯胺酮和地卓西平马来酸盐)均可通过结合谷氨酸的NMDA受体而非竞争性抑制该受体作用,从而诱导精神分裂症的行为学表征[57]。
一些研究也证实了精神分裂症患者脑中的谷氨酸能系统异常。在精神分裂症患者大脑皮层组织中, 突触小体中谷氨酸释放明显缺陷,而使用氟哌啶醇预处理这些组织可部分逆转该作用[58]; 同时,精神分裂症患者前额叶皮层中,谷氨酸再吸收和海人酸受体的结合均增强[59, 60],一定程度上反映了患者谷氨酸突触传递活动的降低。相比之下,患者的壳核、海马体及旁海马皮质中的海人酸受体结合有一定程度降低[61,62],在基底神经节中谷氨酸的重吸收[63]和NMDA受体的表达[64]也有一定程度的降低。这些研究充分证明了谷氨酸传递在精神分裂症患者大脑中发挥了重要作用,且病理下谷氨酸系统的增强和减弱也有强烈的脑区特异性。
3 神经递质检测的技术和器件
目前, 神经递质检测技术的技术和元器件开发已得到了多方面发展[51, 65~69]。在检测递质的电化学方法中,采用伏安法可测定多种自身具有氧化还原特性的神经递质,典型的包括多巴胺、去甲肾上腺素、血清素等; 通过酶在电极表面与递质发生反应并用电化学方法检测该过程的酶传感器也得到了多种应用,包括谷氨酸、乙酰胆碱等递质的检测; 而为了在各种生物实验设定下得到脑组织内递质浓度分布的空间信息,细胞表达的荧光探针结合成像技术也已可探测多种神经递质分子; 综合考虑检测的时间分辨率、样品使用量、电极生产的稳定性和可重复性等方面的需求,进一步使用微流控器件和场效应管(Field effect transistor,FET)等器件的开发也已取得了多方面应用。本节将就这些方面分别举例说明神经递质分子检测技术和器件的研究展。
3.1 伏安法测定递质浓度
在多种电化学技术中,快速循环伏安法(Fast scan cyclic voltammetry, FSCV)被用于神经递质分子监测,也是在体神经递质浓度监测较成熟的技术。这种技术可在单个位点上准确测量一些种类神经递质分子的浓度变化,并且时空精度高于微透析等测量方式。除FSCV外,差分脉冲伏安法(Differential pulse voltammetry, DPV)和方波伏安法(Square wave voltammetry, SWV)等方法在递质检测中也发挥了重要作用。关于FSCV测定递质浓度的生物传感器已有大量研究报道,且多数FSCV方法采用碳纤维电极[70]。同时,近期还有一些研究基于新材料的循环伏安法测定神经递质浓度,如一种免疫传感器表面采用了多巴胺单克隆抗体修饰的磁性纳米颗粒[10]。在磁性纳米颗粒的表面附着了碳纳米管(Carbon nanotubes, CNTs),将这种复合物颗粒通过磁力吸附在金电极的表面上,可采用循环伏安法测定多巴胺的浓度,测定范围可达到780 pmol/L~50 mmol/L,检测限(Limit of detection, LOD)可达到120 nmol/L。另一项研究中,通过碳纤维电极测定血小板悬浮液分泌的血清素[71],采用了FSCV方法,可得到较好的时间分辨率和灵敏度,并且可通过循环伏安法针对性研究血清素的氧化还原特性。近期开发的一种无线传输的实时电化学传感器也可通过FSCV方法测定血清素的浓度[72],该研究使用的1000 V/s的N型快速线性扫描电流(+0.2 V到0.1 V,再到+0.2 V)可在背侧中缝核的脑片上检测电刺激诱发释放的血清素浓度,这种传感器可检测到10 mmol/L的血清素。
除FSCV外,还有多种伏安法可用于生物传感器。如血清素传感器利用分子与配体相互作用的检测方式,采用金电极表面的自组装巯甲丙脯氨酸和苯硫醇单分子层[73]。血清素分子可被选择性强吸附在电极表面,从而检测到阳极溶出电流或观察到氧化还原探针的电荷转移电阻升高。此研究中差分脉冲阳极溶出伏安法和电化学阻抗谱检测血清素的LOD可分别达到28 nmol/L和1.2 nmol/L。
3.2 酶传感器
如图2A所示,采用计时电流法的酶传感器可测定谷氨酸等多种分子的浓度[74], 但酶的制备且活性易损, 也有研究者以如图2B中的核酸适配体制备无酶传感器[75]。 又如在金电极表面修饰谷氨酸氧化酶(Glutamate oxidase,GluOx)、羧基化多壁碳纳米管(Carboxylated multi walled carbon nanotubes, COOH MWCNTs)、金纳米颗粒和壳聚糖的电极。 实验发现这种电极可在5~500 mmol/L范围内测定谷氨酸的浓度,LOD可达到1.6 mmol/L[76]。多种基于胆碱氧化酶、胆碱酯酶的乙酰胆碱生物传感器也可用于快速测定乙酰胆碱的浓度[77],这种传感器常将酶固定在各种膜层材料或固相表面上,采用计时电流法测定乙酰胆碱的浓度。另一种胆碱和乙酰胆碱的酶传感器采用了碳纤维表面电聚合的聚邻苯二胺膜层[78],同时在膜层固定乙酰胆碱酯酶和胆碱氧化酶,随后沉积的是CNT和萘酚膜。这种计时电流法传感器可检测胆碱和乙酰胆碱的浓度,LOD可达45 nmol/L。为了提高传感器的灵敏度,酶传感器需要达到近似于单分子层的覆盖条件[79]。降低膜层的厚度可有效地提高酶传感器的时间精度,因为这种结构可有效地降低检测对象分子扩散到达酶层的时间。这项研究中,纳米薄层石墨烯修饰的碳纤维电极表面以摩尔比1∶10固定了胆碱酯酶和胆碱氧化酶,这样可达到最佳的酶反应效率。计时电流法可在10 μmol/L~4 mmol/L范围内线性检测乙酰胆碱浓度,LOD为10 μmol/L。另一种基于14~70 nm微管结构的乙酰胆碱传感器,可采用计时电流法测定250 μmol/L~6 mmol/L的乙酰胆碱浓度[80],其LOD为205 μmol/L。
尽管循环伏安法已被成功用于各種在体和离体的多巴胺浓度测定,也有研究通过在碳纤维电极表面交联固定的酪氨酸酶开发针对多巴胺检测的酶传感器[81]。在反应中,酪氨酸酶可催化多巴胺和o 多巴醌的转化,从而实现电流法测定多巴胺浓度, 在10 nmol/L~220 mmol/L浓度范围内线性关系良好,LOD为1 nmol/L。
3.3 在体分子遗传荧光探针
光学成像神经递质检测技术是一种可通过检测递质与荧光探针结合发光强弱变化测定分子浓度的方法[82],图2C显示了一种谷氨酸荧光探针的原理[82]。与微透析、电流记录、电化学检测技术相比,这是一种相对无创且高通量的检测方式。为了通过光学成像方法检测某种神经递质分子的动态变化,这种检测荧光蛋白需要满足以下条件:必须直接、特异地与目标分子发生结合,并导致蛋白构型的改变; 荧光强度必须随着这种蛋白构型的改变而发生变化。
目前,已开发的具有一定代表性的递质分子荧光探针已可在多种性能方面实现相对较好的递质浓度检测,如γ 氨基丁酸荧光探针传感器。γ 氨基丁酸 Snifit的Kd=100 μmol/L,荧光强度变化(ΔF/F0)可达到0.5,时间常数Ton=1.5 s,Toff=2.8 s[83]; 谷氨酸荧光探针传感器iGluSnFR的Kd=4.9 μmol/L, 荧光强度变化可达到1.03,时间常数为92 ms[8]; 多巴胺荧光探针传感器D2 CNiFER的Kd=2.5 nmol/L, 荧光强度变化可达到0.57,时间常数小于7 s[84]; 乙酰胆碱荧光探针传感器GACh的Kd=0.78 μmol/L,荧光强度变化可达到0.9,时间常数为280 ms[85]; 去甲肾上腺素荧光探针传感器a2AAR cam的Kd=17 nmol/L,荧光强度变化可达到
0.05,时间常数为40 ms[86]。
总之,尽管很多种神经递质荧光探针仍然在信噪比和时间精度方面仍有一定局限性,但这些可通过遗传操作表达的神经递质传感器可提供较好的分子选择性、空间精度和细胞特异性,而对于在体研究,这些属性均具有重要作用。
3.4 微流控器件
微流控器件在充分发挥电极材料优势并可重复生产等方面具有独特的优势,因此在近年受到越来越多的重视。NO在调控突触传递方面发挥着重要作用,通过在金/氧化铟锡电极表面沉积一层催化NO转化成N2的铁氰化物镀层作为工作电极而构建的微流控装置,可用于测定NO的浓度,进一步通过透气膜层增加传感器对NO的选择性,这种传感器对NO的LOD可达到1 nmol/L[87]。在中枢神经系统中抗坏血酸可有效地对抗谷氨酸诱导的神经毒性作用,并且星型胶质细胞可通过抗坏血酸和谷氨酸分子的胞内外跨膜交换而实现降低胞外谷氨酸浓度[88,89]。针对在体抗坏血酸浓度的检测,一种单通道的微流控期间可达到很好的检测效果[90]。这种方法采用了氧化铟锡玻璃上修饰CNT的方式制备工作电极,从而促进抗坏血酸在电极表面的氧化。通过这种技术观察到,在脑缺血的过程中, 抗坏血酸浓度的逐渐降低; 而在脑缺血再灌注过程中, 抗坏血酸浓度明显升高。另外,由于Mg2+可通过阻断钙通道的方式对NMDA受体发挥重要的调控作用[91~93],针对抗坏血酸和Mg2+实现同步检测的新型微流控器件技术可能在谷氨酸能传导的研究中发挥起到重要作用。
通过光蚀刻技术制备的微流控芯片通常具有灵敏度高、快速响应、检测限低且线性检测范围较好的优势, 可应用于生物传感器[73]。如一种基于计时电流法的双电极微流控芯片, 采用在芯片上固定乙酰胆碱酯酶和胆碱氧化酶的方式检测乙酰胆碱的浓度, 在优化的基底材料上分别固定这两种酶, 制成的传感器可提供更宽的动态区间,更低的检测限及优良的稳定性和可重复性,因此可提高乙酰胆碱传感器的检测效率[94]。
3.5 场效应管传感器
由于神经递质的释放通常发生在毫秒量级,基于场效应管的快速生物传感器可在研究中起到独特的重要作用,因而这些场效应管传感器在生物传感器领域也逐渐引起关注,如一种SWCNT场效应管传感器采用了表达m型乙酰胆碱受体的大肠杆菌修饰,这样可提高乙酰胆碱检测的灵敏性和选择性。这种传感器对乙酰胆碱的LOD可达到100 pmol/L[95]。
另外一项研究中,使用多晶硅线场效应管制成的多巴胺传感器,可在fmol/L级别检测多巴胺的浓度,并且这种传感器的制备可通过现有的商用半导体制造技术实现,因此可能迅速实现稳定生产[96]。
4 神经递质检测的电极材料
很多种新型电极材料已被用于制作检测神经递质的生物传感器,不同材料在各种递质的检测中发挥各自的独特作用。其中,金属纳米结构材料可通过模仿酶活性中心的方式, 实现递质的无酶检测,而核酸适体材料则可通过测定构像改变后氧化还原电流的变化或电极阻抗变化的方式实现递质的无酶检测。这些不依赖电极表面固定酶的生物传感器材料既可解决酶作为蛋白质可能变性失活的问题,又可大幅度降低传感器的制作成本,从而方便技术的推广应用。但体外传感器技术取得的检测效果在转移到体内应用时都面临多种问题,如电极表面的细胞和蛋白质生物污损更加复杂多样等。
4.1 碳纳米管和石墨烯
采用在电极表面镀导电膜层的方式可一定程度上改进对多巴胺的检测性能[97]。例如,带正电的多巴胺分子和带负电的卟啉修饰石墨烯电极间可通过分子间芳环作用和静电力加速电子转移,图3A展示了石墨烯分子的常见结构形态。相比之下,带负电的干扰分子(如抗坏血酸和尿酸分子)可被带负电的卟啉电极表面抑制,这种电极对多巴胺的检测LOD可达到10 nmol/L。 Prasad等[98]以整合了CNT的聚合材料网络构成中的肾上腺素分子作为模板分子,在石墨电极表面使用这种复合材料制成分子印迹传感器电极,采用差分脉冲阳极溶出伏安法(Differential pulse anodic stripping voltammetry, DPASV)测定肾上腺素的浓度,线性范围在0.5~32 nmol/L之间, LOD可达到10.9 nmol/L。
在血清素检测方面, 碳纳米材料也有多种应用。例如, Sun等[99]采用CNT和离子液体的复合电极材料,可同时检测血清中的血清素和多巴胺的浓度,血清素的LOD可低至8 nmol/L。然而, 由于多种生物组织液中都普遍存在尿酸, 而尿酸在这种电极上的干扰信号可达到10倍以上,因此限制了其应用。Han等[100]在卟啉修饰的玻碳电极上采用还原的氧化石墨烯材料检测血清素, LOD可低至4.9 nmol/L。 但由于缺乏实际样品的测试,尚不确定该电极是否可能用于临床样本检测。由于低阻抗和大的反应面积, 碳纳米管在多种递质的检测中均已展现出一定优势,图3B展示了一种MWCNT修饰的电极可实现的大比表面积多孔三维结构[101]。Goyal等[102]采用MWCNT修饰电极,基于方波脉冲法测定了吸烟人群血液中的肾上腺素,相较于未修饰的电极, MWCNT修饰电极可对肾上腺素氧化发挥催化活性, 进而升高电流并降低氧化电位, LOD可达到0.15nmol/L。
Xiang等[103]基于在碳纖维表面垂直排布的CNT阵列检测了大鼠脑中抗坏血酸的浓度; 并通过在这些CNT阵列表面沉积铂层的方法,制备了可检测脑缺血/再灌注过程中氧气浓度变化的传感器[104]。
4.2 金属纳米镀层和纳米颗粒
大量研究证明, 在电极表面合成和沉积金属纳米材料是一种相对简单的提高生物传感器性能的方式[105,106]。 垂直排列的镍纳米线和镀铂镍纳米线修饰的电极可在不依赖酶的条件下实现谷氨酸浓度的电化学检测。这些检测电极总体检测较灵敏并且比酶电极成本低,利于推广(图3C)[107]。研究表明,这种材料制成的电极可检测浓度低于8 mmol/L的谷氨酸,而镍纳米线和镀铂镍纳米线修饰电极的LOD可分别达到68和83 μmol/L[108]。
另一种方式是基于仿生的原理, 通过金属材料实现类似酶催化作用的电极材料[109],因为多种酶的催化中心都通过不同的金属离子实现催化作用,如使用铜离子复合物和银纳米颗粒修饰的玻碳电极可制备多巴胺传感器。这种铜离子复合物材料可模仿酪氨酸酶的循环催化作用,特别是用双氧水预处理电极的条件下可提高多巴胺检测的峰电流值,从而提高检测性能。另外,金属纳米颗粒在递质传感器中也有重要的应用,图3D展示了一种金纳米颗粒的结构特点[76], 这种纳米颗粒制成都的复合材料可有效检测谷氨酸浓度。研究发现,银纳米颗粒对多巴胺的电催化效果远强于未修饰的普通金属电极。而通过在玻碳电极表面修饰氧化镍纳米颗粒和CNT材料, 并采用针对肾上腺素的双十六烷基磷酸盐膜覆盖的方式制备的传感器也可通过差分脉冲伏安法测定肾上腺素的浓度[110],检测范围为0.3~9.5 μmol/L, LOD可达到82 nmol/L。
4.3 核酸适体
如图2B所示,除了使用酶检测神经递质的方法,也可用通过检测核酸适体的构像改变方式实现递质分子的检测[12,111]。即如图中展示的多巴胺核酸适体传感器[75]。 针对各种神经递质分子的核酸适体均可通过指数富集的配体系统进化(Systematic evolution of ligands by exponential Enrichment,SELEX)过程筛选出来[112],为传感器的开发提供了便利条件。 人工合成的适体分子易于化学修饰,从而提高选择性、响应时间和稳定性等多种性质[113, 114]。通过适体5'端的巯基结合到金电极上,且在3'端结合亚甲基蓝的方式,适体可在目标分子存在的时候改变构像从而拉近亚甲基蓝分子和电极的距离,检测电流变化即可测定目标分子浓度[115],图中ET表示电子传递(Electron transfer)。此技术响应时间为45~100 s,亲和力可达微摩尔级, 并且可实现生理浓度下的分子检测,而且该技术也被证实可在体检测4.5 h以上。
Farjami等[116]通过计时电流法测定带负电的核酸适体和带正电的半胱氨酸修饰电极之间的相互作用,实现多巴胺浓度的测定,检测范围为100 nmol/L~5 mmol/L,采用电极表面镀萘酚膜的方式,抗坏血酸和尿酸的干扰均可基本排除。通过调控适体与电极表面结合的静电力[74],还可进一步提高多巴胺分子的亲和力,在最佳状态下, 适体解离常数可达到(0.12±0.01 )mmol/L。
然而, 核酸适体传感器也面临一些挑战,由于缺乏稳定的电极表面膜层, 可通过限制扩散而筛选带电大分子,这类电极通常不能用于测定大分子浓度。但这些传感器通常都可以测定药用小分子在特定组织内的浓度。这些核酸适体有时会与不同的待测药物分子反应,但对于药用分子浓度测定而言, 这些限制通常不是决定性因素,因为在一般情况下, 这种药物分子在体内不存在结构过度类似的分子,但这些传感器测定神经系统内源性生物标记物分子可能会遇到困难,因为在组织内部这些分子的代谢过程中常有结构近似的分子,可能需要通过更多轮次的SELEX正向/负向选择, 提高适体的特异性。另外,核酸适体等具备传感功能的分子,由于其结构基础与生物体本身的遗传物质DNA相同,可能受到体内核酸内切酶等多种酶的催化降解作用。
5 神经递质检测技术的局限与展望
目前,递质检测技术受到了越来越多的重视,特别是与电生理和光遗传等技术联用的可能性吸引了神经科学和工程材料领域的共同关注。目前,基于导电聚合物和CNT等修饰材料的电极已被成功用于在体电生理研究[9,108,117~120],神经电极修饰的COOH CNT一方面可用于共价连接特定的修饰分子(如酶分子),从而实现在体传感器的制备,也可用于快速循环伏安法测定多巴胺等递质[121]; 另一方面,一些柔性神经电极材料也引起越来越多的关注[122],而这些电极的聚合物导电尖端或者绝缘层也可能与在体电化学传感器联用,实现慢性植入下生物相容性更好的电化学检测。一些新型的神经界面电极材料(如碳纳米管束电极等)也可实现在体的分子检测[123,124]。
然而,电生理和电化学检测技术由于互相之间的电流渗漏而难以直接在系统中整合联用,而开发异步分时采集电化学和电生理信号的技术相对易于实现[66]。一些实验已证实,可在黑质中进行电生理记录, 并在新纹状体中采用电化学方法检测下游的多巴胺浓度[125]。也有一些研究尝试采用同一个电极阵列上不同位点分别采集电生理信号和电化学信号的方法,当记录位点距离为200 μm时,计时电流法对场电位和单神经元活动的影响较小[126]。
一些电化学修饰材料除了可降低神经电极的阻抗,也可用于精准控制药物释放 [127],这项技术可与电化学传感器联用, 而实现对某种或某些神经递质的闭环调控。 这种对神经递质闭环调控的概念逐渐受到研究者的重视[128]。
另一方面,光遗传技术在神经科学中的应用日趋广泛,如本能恐惧行为的皮层下通路[129, 130]与癫痫[131]、老年痴呆[132]等疾病的研究。目前,FSCV技术与光遗传技术的结合已在社交孤立模型的环路机制研究等方面取得了一些重要进展[133]。但光遗传技术与在体传感器的结合仍处于应用早期,有广阔的发展空间[134]。由于光遗传技术使用的光纤也可用于在体荧光测定递质浓度,两者的结合可能为局部环路的闭环递质浓度检测与调控技术提供新的解决方案。 另外,由于光遗传技术本身并不受电流的影响,因此光遺传学调控神经元活动可与电化学递质浓度检测结合起来,从而避免电生理方法等与电化学联用检测递质浓度过程中存在的各种障碍。
尽管面临诸多机遇,但由于大脑的复杂性,在不破坏整体组织完整性的前提下检测某种特定递质浓度,仍要面对多种干扰物分子同时游离在检测电极周围的复杂情况,因而在体传感器也面临着许多挑战:(1)电极尺寸需足够小,从而能在尽可能不破坏大脑本身组织结构的情况下测定递质浓度,而小电极尺寸也为具有更高空间精度的递质检测提供了可能; (2)在体递质传感器必须满足快速响应和灵敏度高等特征,因而目前的各种技术均面临一定挑战,而高性能传感器中,有的器件结构复杂、精密度高,会影响成品的优良率,且会提升在体应用中丧失功能的风险; (3)高选择性对分析技术十分重要,但是体内分子种类复杂多样,因此生物传感器检测信号的解读需格外慎重,多种在体检测信号的联用和交叉检查可能提供更具参考性的信息,但可能显著增加难度和开发周期; (4) 某些用于修饰的纳米材料等对生物组织有潜在毒性,需谨慎考虑并测定这些因素的影响程度,特别是对潜在的在体应用; (5)蛋白质和细胞组织在电极表面的贴附会导致电极的生物污损或生物毒性效应,特别是利用多肽和核酸适体等分子的电极材料需慎重考虑内源性酶对这些分子的潜在降解,及其对传感器在体寿命的影响。
通过递质传感器技术的改进、与其它神经技术的联用以及针对在体环境设计与测试等方面的方法学推进,递质检测技术有望在阐明神经系统中各种疾病发生的机制和提供针对性治疗方案等方面发挥更重要的作用。
References
1 Cooper J R, Bloom F E, Roth R H, The Biochemical Basis of Neuropharmacology. Oxford University Press, 1996: 1-528
2 Meldrum B S. J. Nutrition, 2000, 130(4 SUPPL.): S1007-S1015
3 Marvin J S, Borghuis B G, Tian L, Cichon J, Harnett M T, Akerboom J, Gordus A, Renninger S L, Chen T W, Bargmann C I, Orger M B, Schreiter E R, Demb J B, Gan W B, Hires S A, Looger L L. Nat. Methods, 2013, 10(2): 162 170
4 Du Z J, Luo X, Weaver C L, Cui X T. J. Mater. Chem. C, 2015, 3(25): 6515-6524
5 Baldrich E, Muoz F X. Anal. Chem., 2011, 83(24): 9244-9250
6 Lu Y, Li Y, Pan J, Wei P, Liu N, Wu B, Cheng J, Lu C, Wang L. Biomaterials, 2012, 33(2): 378-394
7 Taylor I, Du Z J, Bigelow E, Eles J, Horner A R, Catt K A, Weber S, Jamieson B, Cui X T. J. Mater. Chem. B, 2017, 5(13): 2445-2458
8 Silva P N, Green B J, Altamentova S M, Rocheleau J V. Lab Chip, 2013, 13(22): 4374-4384
9 P S Booth M, Kosmopoulou M, Poirel L, Nordmann P, Spencer J. Plos One, 2015 10(10): e0140059
10 Bradford H F. Prog. Neurobiol., 1995, 47(6): 477-511
11 Treiman D M. Epilepsia, 2001, 42(s3): 8-12
12 Bradford H, Dodd P. Biochem. Neurol. Dis., 1976: 114-167
13 Roberts E. Annu. Neurol., 1984, 16(1S): S77 S89
14 van Gelder N M. Neurochem. Res., 1987, 12(2): 111-119
15 Gjessing L R, Gjesdahl P, Sjaastad O. J.Neurochem., 1972, 19(7): 1807-1808
16 Ribak C E. Research Publications Association for Research in Nervous and Mental Disease, 1983, 61: 109-130
17 Ribak C E. Brain Res., 1985, 326(2): 251-260
18 Babb T L, Pretorius J K, Kupfer W R, Crandall P H. J. Neurosci., 1989, 9(7): 2562-2574
19 Callahan P M, Paris J M, Kathryn A C, Shinnick Gallagher P. Brain Res., 1991, 555(2): 335-339
20 Lscher W, Schwark W S. Brain Res., 1987, 420(2): 385-390
21 Lloyd K, Bossi L, Morselli P, Rougier M, Loiseau P, Munari C. Epilepsy GABA Receptor Agonists: Basic and Therapeutic Research, 1985: 43-51
22 Rougier A, Lloyd K G, Dartigues J F, Loiseau H, Morselli P L, Loiseau P. Adv. Epileptol., 1989, 17: 131-134
23 Kaura S, Bradford H F, Young A M J, Croucher M J, Hughes P D. J. Neurochem., 1995, 65(3): 1240-1249
24 Hornykiewicz O. Pharmacol. Rev., 1966, 18(2): 925
25 Hornykiewicz O. Fed. Proc., 1973, 32(2): 183-190
26 Hornykiewicz O. In Movement Disorders, edited by Marsden C D, Fahn S (Butterworth Heinemann, 1981: 41-58
27 Fuxe K, Hkfelt T, Ungerstedt U. Int. Rev. Neurobiol., 1970, 3: 93-126
28 Moore R Y, Bloom F E. Annu. Rev. Neurosci., 1978, 1(1): 129-169
29 Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. J. Neurol. Sci., 1973, 20(4): 415-455
30 Lloyd K G, Davidson L, Hornykiewicz O. J. Pharmacol. Exp. Ther., 1975, 195(3): 453
31 Price K S, Farley I J, Hornykiewicz O. Adv. Biochem. Psychopharmacol., 1978, 19: 293-300
32 Hazell A S. Neurochem. Int., 2007, 50(7): 941-953
33 Rao V L R, Bowen K K, Dempsey R J. Neurochem. Res., 2001, 26(5): 497-502
34 Rothstein J D, Dykes Hoberg M, Pardo C A, Bristol L A, Jin L, Kuncl R W, Kanai Y, Hediger M A, Wang Y, Schielke J P, Welty D F. Neuron, 1996, 16(3): 675-686
35 Yeh T H, Hwang H M, Chen J J, Wu T, Li A H, Wang H L. Neurobiol. Dis., 2005, 18(3): 476-483
36 Giffard R G, Monyer H, Choi D W. Brain Res., 1990, 530(1): 138-141
37 Swanson R A, Farrell K, Stein B A. Glia, 1997, 21(1): 142-153
38 Lees G J. Brain Res. Rev., 1991, 16(3): 283-300
39 Longuemare M C, Swanson R A. J. Neurosci. Res., 1995, 40(3): 379-386
40 Seki Y, Feustel P J, Keller Jr R W, Tranmer B I, Kimelberg H K. Stroke, 1999, 30(2): 433-440
41 Szatkowski M, Barbour B, Attwell D. Nature, 1990, 348(6300): 443-446
42 Dronne M A, Grenier E, Dumont T, Hommel M, Boissel J P. Brain Res., 2007, 1138(1): 231-242
43 Alberdi E, Sánchez Gómez M V, Marino A, Matute C. Neurobiol. Dis., 2002, 9(2): 234-243
44 Li S, Mealing G A, Morley P, Stys P K. J. Neurosci., 1999, 19(14): RC16
45 Matute C, Sánchez Gómez M V, Martínez Millán L, Miledi R. Proc. Natl. Acad. Sci. USA, 1997, 94(16): 8830-8835
46 McDonald J W, Althomsons S P, Hyrc K L, Choi D W, Goldberg M P. Nat. Med., 1998, 4(3): 291-297
47 Sánchez Gómez M V, Matute C. Neurobiol. Dis., 1999, 6(6): 475-485
48 Fern R, Ransom B R, Waxman S G. J. Neurophysiol., 1995, 74(1): 369-377
49 Stys P K, Ransom B R, Waxman S G, Davis P K. Proc. Natl. Acad. Sci. USA, 1990, 87(11): 4212-4216
50 Wolf J A, Stys P K, Lusardi T, Meaney D, Smith D H. J. Neurosci., 2001, 21(6): 1923-1930
51 Sangubotla R, Kim J. TrAC Trend. Anal. Chem., 2018, 105: 240-250
52 Caraci F, Nicoletti F, Copani A. Curr. Opin. Pharmacol., 2018, 38: 1-7
53 Si B, Song E. Chemosensors, 2018, 6(1): 1-24
54 Tsai G, Passani L A, Slusher B S, Carter R, Baer L, Kleinman J E, Coyle J T. JAMA Psychiat., 1995, 52(10): 829-836
55 Deutsch S I, Mastropaolo J, Schwartz B L, Rosse R B, Morihisa J M. Clin. Neuropharmacol., 1989, 12(1): 1-13
56 Javitt D C, Zukin S R. Am. J. Psychiat., 1991, 148(10): 1301-1308
57 Krystal J H, Karper L P, Seibyl J P, Freeman G K, Delaney R, Bremner J D, Heninger G R, Bowers M B, Charney D S. Arch. Gen. Psychiat., 1994, 51(3): 199-214
58 Sherman A D, Davidson A T, Baruah S, Hegwood T S, Waziri R. Neurosci. Lett., 1991, 121(1 2): 77-80
59 Deakin J F W, Slater P, Simpson M D C, Gilchrist A C, Skan W J, Royston M C, Reynolds G P, Cross A J. J. Neurochem., 1989, 52(6): 1781-1786
60 Nishikawa T, Takashima M, Toru M. Neurosci. Lett., 1983, 40(3): 245-250
61 Kerwin R, Patel S, Meldrum B. Neuroscience, 1990, 39(1): 25-32
62 Kerwin R W, Patel S, Meldrum B S, Czudek C, Reynolds G P. Lancet, 1988, 331(8585): 583-584
63 Simpson M D C, Slater P, Royston M C, Deakin J F W. Psychiat. Res., 1992, 42(3): 273-282
64 Kornhuber J, Mack Burkhardt F, Riederer P, Hebenstreit G F, Reynolds G P, Andrews H B, Beckmann H. J. Neural Transm., 1989, 77(2 3): 231-236
65 Hasanzadeh M, Shadjou N, Guardia M D. TrAC Trend. Anal. Chem., 2017, 86: 107-121
66 Wang Y, Mao L. Electroanalysis, 2016, 28(2): 265-276
67 Xiao T, Wu F, Hao J, Zhang M, Yu P, Mao L. Anal. Chem., 2017, 89(1): 300-313
68 Wu F, Yu P, Mao L. Chem. Soc. Rev., 2017, 46(10): 2692-2704
69 Ganesana M, Lee S T, Wang Y, Venton B J. Anal. Chem., 2017, 89(1): 314-341
70 zel R E, Hayat A, Andreescu S. Anal. Lett., 2015, 48(7): 1044-1069
71 Ge S, Woo E, White J G, Haynes C L. Anal. Chem., 2011, 83(7): 2598-2604
72 Griessenauer C J, Chang S Y, Tye S J, Kimble C J, Bennet K E, Garris P A, Lee K H. J. Neurosurg., 2010, 113(3): 656-665
73 Mozaffari S A, Chang T, Park S M. Biosens. Bioelectron., 2010, 26(1): 74-79
74 Rahman M M, Asiri A M. Anal. Methods, 2015, 7(22): 9426-9434
75 lvarez Martos I, Campos R, Ferapontova E E. Analyst, 2015, 140(12): 4089-4096
76 Batra B, Pundir C S. Biosens. Bioelectron., 2013, 47: 496-501
77 Khan A, Khan A A P, Asiri A M, Rub M A, Azum N, Rahman M M, Khan S B, Ghani S A. Appl. Biochem. Biotech., 2013, 169(6): 1927-1939
78 Khan A, Ghani S A. Biosens. Bioelectron., 2012, 31(1): 433-438
79 Keighron J D, Wigstrm J, Kurczy M E, Bergman J, Wang Y, Cans A S. ACS Chem. Neurosci., 2015, 6(1): 181-188
80 Colombo M L, Sweedler J V, Shen M. Anal. Chem., 2015, 87(10): 5095-5100
81 Njagi J, Chernov M M, Leiter J C, Andreescu S. Anal. Chem., 2010, 82(3): 989-996
82 Wang H, Jing M, Li Y. Curr. Opin. Neurobiol., 2018, 50: 171-178
83 Masharina A, Reymond L, Maurel D, Umezawa K, Johnsson K. J. Am. Chem. Soc., 2012, 134(46): 19026-19034
84 Muller A, Joseph V, Slesinger P A, Kleinfeld D. Nat. Methods, 2014, 11(12): 1245-1252
85 Jing M, Zhang P, Wang G, Feng J, Mesik L, Zeng J, Jiang H, Wang S, Looby J, Guagliardo N A, Langma L W, Lu J, Zuo Y, Talmage D A, Role L W, Barret P Q, Zhang L I, Luo M, Song Y, Zhu J J, Li Y. Nat. Biotechnol., 2018, 36(8): 726-737
86 Vilardaga J P, Bünemann M, Krasell C, Castro M, Lohse M J. Nat. Biotechnol., 2003, 21(7): 807-812
87 Cha W, Tung Y C, Meyerhoff M E, Takayama S. Anal. Chem., 2010, 82(8): 3300-3305
88 Wilson J X, Peters C E, Sitar S M, Daoust P, Gelb A W. Brain Res., 2000, 858(1): 61-66
89 Portugal C C, Miya V S, Calaza K D C, Santos R A M, Paes De Carvalho R. J. Neurochem., 2009, 108(2): 507-520
90 Lin Y, Lu X, Gao X, Cheng H, Ohsaka T, Mao L. Electroanalysis, 2013, 25(4): 1010-1016
91 Silver I A, Erecńska M. J. Gen. Physiol., 1990, 95(5): 837-866
92 Choi D W, Rothman S M. Annu. Rev. Neurosci., 1990, 13: 171-182
93 Bano D, Nicotera P. Stroke, 2007, 38(2 PART 2): 674-676
94 Akhtar M H, Hussain K K, Gurudatt N G, Shim Y B. Biosens. Bioelectron., 2017, 98: 364-370
95 Kim B, Song H S, Jin H J, Park E J, Lee S H, Lee B Y, Park T H, Hong S. Nanotechnology, 2013, 24: 375302
96 Lin C H, Hsiao C Y, Hung C H, Lo Y R, Lee C C, Su C J, Lin H C, Ko F H, Huang T Y, Yang Y S. Chem. Commun., 2008, (44): 5749-5751
97 Wu L, Feng L, Ren J, Qu X. Biosens. Bioelectron., 2012, 34(1): 57-62
98 Prasad B B, Prasad A, Tiwari M P, Madhuri R. Biosens. Bioelectron., 2013, 45(1): 114-122
99 Sun Y, Fei J, Hou J, Zhang Q, Liu Y, Hu B. Microchim. Acta, 2009, 165(3 4): 373-379
100 Han H S, Lee H K, You J M, Jeong H, Jeon S. Sens. Actuators B, 2014, 190: 886-895
101 Kozai T D Y, Catt K, Du Z, Na K, Srivannavit O, Haque R M, Seymour J, Wise K D, Yoon E, Cui X T. IEEE T. Biomed. Eng., 2016, 63(1): 111-119
102 Goyal R N, Bishnoi S. Electrochim. Acta, 2011, 56(6): 2717-2724
103 Xiang L, Yu P, Hao J, Zhang M, Zhu L, Dai L, Mao L. Anal. Chem., 2014, 86(8): 3909-3914
104 Xiang L, Yu P, Zhang M, Hao J, Wang Y, Zhu L, Dai L, Mao L. Anal. Chem., 2014, 86(10): 5017-5023
105 Doria G, Conde J, Veigas B, Giestas L, Almeida C, Assuno M, Rosa J, Baptista P V. Sensors, 2012, 12(2): 1657-1687
106 Daniel M C, Astruc D. Chem. Rev., 2004, 104(1): 293-346
107 Jamal M, Hasan M, Mathewson A, Razeeb K M. Biosens. Bioelectron., 2013, 40(1): 213-218
108 Labib M, Sargent E H, Kelley S O. Chem. Rev., 2016, 116(16): 9001-9090
109 Sanghavi B J, Mobin S M, Mathur P, Lahiri G K, Srivastava A K. Biosens. Bioelectron., 2013, 39(1): 124-132
110 Figueiredo Filho L C S, Silva T A, Vicentini F C, Fatibello Filho O. Analyst, 2014, 139(11): 2842-2849
111 Arroyo Currás N, Somerson J, Vieira P A, Ploense K L, Kippin T E, Plaxco K W. Proc. Natl. Acad. Sci. USA, 2017, 114(4): 645-650
112 Meng H M, Liu H, Kuai H, Peng R, Mo L, Zhang X B. Chem. Soc. Rev., 2016, 45(9): 2583-2602
113 Yang L, Qing Z, Liu C, Tang Q, Li J, Yang S, Zheng J, Yang R, Tan W. Anal. Chem., 2016, 88(18): 9285-9292
114 Shi H, He X, Cui W, Wang K, Deng K, Li D, Xu F. Anal. Chim. Acta, 2014, 812: 138-144
115 Ferguson B S, Hoggarth D A, Maliniak D, Ploense K, White R J, Woodward N, Hsieh K, Bonham A J, Eisenstein M, Kippin T E, Plaxco K W, Soh H T. Sci. Transl. Med., 2013, 5(213): 213ra165
116 Farjami E, Campos R, Nielsen J S, Gothelf K V, Kjems J, Ferapontova E E. Anal. Chem., 2013, 85(1): 121-128
117 Kolarcik C L, Catt K, Rost E, Albrecht I N, Bourbeau D, Du Z, Kozai T D Y, Luo X, Weber D J, Cui X T. J. Neural Eng., 2015, 12(1): 016008
118 Kozai T D Y, Du Z, Gugel Z V, Smith M A, Chase S M, Bodily L M, Caparosa E M, Friedlander R M, Cui X T. J. Neurosci. methods, 2014, 242C: 15-40
119 Lu Y, Li T, Zhao X, Li M, Cao Y, Yang H, Duan Y Y. Biomaterials, 2010, 31(19): 5169-5181
120 Lu Y, Wang D, Li T, Zhao X, Cao Y, Yang H, Duan Y Y. Biomaterials, 2009, 30(25): 4143-4151
121 Taylor I M, Robbins E M, Catt K A, Cody P A, Happe C L, Cui X T. Biosens. Bioelectron., 2017, 89(Pt 1): 400-410
122 Du Z J, Kolarcik C L, Kozai T D Y, Luebben S D, Sapp S A, Zheng X S, Nabity J A, Cui X T. Acta Biomaterialia, 2017, 53: 46-58
123 Schmidt A C, Wang X, Zhu Y, Sombers L A. ACS Nano, 2013, 7(9): 7864-7873
124 Lugo Morales L Z, Loziuk P L, Corder A K, Toups J V, Roberts J G, McCaffrey K A, Sombers L A. Anal. Chem., 2013, 85(18): 8780-8786
125 Kuhr W G, Wightman R M, Rebec G V. Brain Res., 1987, 418(1): 122-128
126 Johnson M D, Franklin R K, Gibson M D, Brown R B, Kipke D R. J. Neurosci. Methods, 2008, 174(1): 62-70
127 Du Z J, Bi G Q, Cui X T. Adv. Funct. Mater., 2017, 28(12): 1703988
128 Belle A M, Owesson White C, Herr N R, Carelli R M, Whiteman R M. ACS Chem. Neurosci., 2013 4(5): 761-771
猜你喜欢
中国典型病例大全(2022年7期)2022-04-22
中国典型病例大全(2022年7期)2022-04-22
中国典型病例大全(2022年7期)2022-04-22
中国典型病例大全(2022年7期)2022-04-22
右江医学(2022年3期)2022-04-17
大众健康(2021年2期)2021-03-09
中国中药杂志(2016年23期)2017-04-07
企业技术开发·下旬刊(2016年11期)2016-12-27
现代经济信息(2016年24期)2016-11-09
