
Diversity scaling of human vaginal microbial communities
2019-10-31 10:51:10WendyLi,Zhan-ShanMa
Zoological Research 2019年6期
DEAR EDITOR,The composition and diversity of the human vaginal microbial community have been investigated intensively due to the diversity-stability relationship (DSR)-based hypothesis for bacterial vaginosis(BV)etiology,which was first proposed in the 1990s and has received renewed interest in recent years.Nevertheless,diversity changes(scaling)across individuals in a cohort or population have not yet been addressed,which is significant both theoretically and practically. Theoretically,biodiversity scaling is the core of biogeography, and practically, inter-subject heterogeneity is critical for understanding the etiology and epidemiology of human microbiome-associated diseases such as BV.Here we applied the diversity-area relationship(DAR),a recent extension to the classic species-area relationship(SAR),to study diversity scaling of the vaginal microbiome by reanalyzing reported data collected from 1107 postpartum women.The model used here characterized the power-law (or its extension)relationships between accrued diversity and areas(numbers of individuals),upon which four biogeographic profiles were thus defined. Specifically, we established the DAR profile(relationship between diversity scaling parameter and sotermed diversity order(q)),similarly pair-wise diversity overlap(PDO)profile,maximal accrual diversity(MAD)profile,and ratio of individual-level to population-level diversity (RIP)profile.These four profiles offer valuable tools to assess and predict diversity scaling (changes) in the human vaginal microbiome across individuals,as well as to understand the dynamics of vaginal microbiomes in healthy women.
The human vaginal microbiome is a complex ecosystem that plays critical roles in maintaining host health.As the first defense of the reproductive tract,the vaginal microbiome is critical for the prevention of opportunistic pathogen colonization and viral infection. For example, endogenous,healthy vaginal microbiota can help protect against HIV infection by activating local and systemic inflammation;however,microbiota associated with BV can also increase susceptibility to HIV infection(Buvé et al.,2014;Petrova et al.,2013).For pregnant women,the vaginal microbiota is not only associated with maternal health but also that of neonates,with the composition of the newly colonized microbiome playing a key role in newborn immunity and metabolic development(Cox et al.,2014;Dominguez-Bello et al.,2010;Olszak et al.,2012;Rutayisire et al.,2016).Furthermore,babies delivered by cesarean section can have a higher risk of metabolic and immune diseases than those delivered vaginally(Dominguez-Bello et al.,2010;Sevelsted et al.,2015;Younes et al.,2018),although Chu et al.(2017)noted that delivery mode does not influence microbiome composition in newborns.Moreover,in pregnancy, vaginal dysbiosis is hypothesized to be a contributor to spontaneous preterm birth(Freitas et al.,2018;Romero et al.,2014a;Stout et al.,2017)and miscarriage(Ralph et al.,1999).
In many healthy women, the vaginal microbiota is dominated by Lactobacillus spp.(Macklaim et al.,2013;Ravel et al.,2011).Several studies(Brotman et al.,2014;Gajer et al.,2012;Ma&Li,2017;Ravel et al.,2011)have confirmed the five major community state types of the vaginal microbiome in adult women,as first identified by Ravel et al.(2011). Four types are dominated by Lactobacillus spp.,including L.iners,L.crispatus,L.gasseri,and L.jensenii.However, 20%-30%of asymptomatic, otherwise healthy individuals lack lactic acid bacteria in their vaginal microbiome, which instead consists of obligate anaerobic bacteria(Ravel et al.,2011,2013).In addition,the frequency of microbiome type varies in different ethnic groups, with those microbiome not dominated by Lactobacillus spp.more commonly found in healthy Hispanic and black women than in Asian or white women(Ma et al.,2012;Ravel et al.,2011).Furthermore,the composition of the vaginal microbiome is dynamic during life and associated with menopause stage(Muhleisen & Herbst-Karlovetz, 2016). Recent research demonstrated the vaginal microbiome of perimenarcheal adolescents to be dominated by Lactobacillus spp.,including L.crispatus,L.iners,L.gasseri,and L.jensenii,similar to that found in reproductive-age women(Hickey et al.,2015).In premenopausal women, the vaginal microbiota is still dominated by L.crispatus and L.iners,but Lactobacillus spp.are often replaced by Streptococcus and Prevotella in the perimenopausal and postmenopausal stages(Brotman et al.,2014).Shifts in vaginal microbiome have also been observed during and after pregnancy. For example, diversity and richness of the vaginal microbiome is lower in pregnant women than in non-pregnant women(Freitas et al.,2017).Furthermore,Romero et al.(2014b)showed that the vaginal microbiome of pregnant women contains a higher abundance of L.vaginalis,L.crispatus,L.gasseri,and L.jensenii,and a lower probability of switching to a Lactobacillus-deficient community.In addition,radical changes in Lactobacillus-poor vaginal communities have been found at delivery,which can persist for up to a year(DiGuilio et al.,2015).
Despite extensive studies on the human vaginal microbiome, what constitutes normal or healthy vaginal microbiota remains unresolved. For example, Doyle et al.(2018)sampled and sequenced the vaginal microbiome of 1 107 rural Malawi women after pregnancy, and found that 75.7% (752/994) of the population were dominated by Gardnerella vaginalis rather than by Lactobacillus spp.,and although L. iners increased with time after delivery, G.vaginalis still dominated for an extended period.In Doyle’s study,both the pregnancy delivery mode and ethnicity also appeared to influence the composition of vaginal microbiome,though all hosts were healthy.
Previous research has revealed that the biodiversity of vaginal microbial communities varies with health status and lifestyle of the host.Nevertheless,existing studies have not addressed diversity scaling(changes)across individuals in a cohort or population. Theoretically, microbiome diversity distribution across individual subjects (i. e., space) is traditionally a focus of microbial biogeography. Practically speaking, understanding the biogeography of the human microbiome can reveal critical information on its characteristics in a cohort setting, which can, in turn,significantly influence studies on the etiology and epidemiology of human microbiome-associated diseases such as inflammatory bowel disease,obesity,and BV.To effectively assess the spatial scaling of human vaginal microbial diversity, we applied the DAR model, which is a recent extension of the classic SAR in biogeography and conservation biology(Bell et al.,2005;Horner-Devin et al.,2004;MacArthur&Wilson,1967;Noguez et al.,2005;Peay et al.,2007;Triantis et al.,2012;Várbíró et al.,2017;Whittaker& Triantis, 2012). SAR is one of the oldest described ecological laws or patterns, whereby species richness increases with increasing sampling area,and can be traced back to the 19th century(Watson,1835).It is still considered one of the most important principles in conservation biology and biogeography. The extensions from SAR to DAR introduced a several important advances including:(1) Expanding species richness (number of species) to general diversity measures in Hill numbers(Chao et al.,2012,2014;Hill,1973),thus making it possible to not only assess the scaling of species richness(numbers),but also scaling of general diversity(e.g.,change in community evenness or dominance).Therefore,the classic SAR is a special case of the more general DAR;(2)The DAR,PDO,MAD,and local regional/global diversity(LRD/LGD)profiles are effective tools for the biogeographic mapping of biodiversity over space(Ma,2018a,2018c,2019).
In this study,we applied DAR modeling and associated biogeographic profiles to investigate the spatial diversity scaling of postpartum vaginal microbial communities across individuals by reanalyzing the large vaginal microbiome dataset originally reported by Doyle et al.(2018).The spatial diversity scaling of the vaginal microbiome revealed heterogeneity among individuals, which could provide an ecological basis for personalized and precise diagnosis and treatment of microbiome-associated diseases,including BV.The biogeographic profiles of the vaginal microbiome also provide tools for explaining the DSR hypothesis for BV etiology from multiple dimensions(Ma&Ellison,2018,2019).
The vaginal microbial dataset (Doyle et al., 2018)reanalyzed in this study consisted of 1 158 vaginal microbiome samples collected from 1 107 rural Malawi women postdelivery.Most samples were collected within the first 20 d of delivery,though some were sampled 5-583 d post-delivery.The V5-V7 hypervariable regions of the 16S rRNA genes were amplified and sequenced under the MiSeq Illumina platform.After quality control,the sequences were clustered into 14 354 operational taxonomic units(OTUs)using QIIME 2.8.6.Samples with less than 2 000 reads were removed,as were OTUs with less than 1 000 reads. After prescreening,1 076 samples and 466 OTUs remained for DAR analysis.In DAR analysis,the number of each OTU read is equivalent to the population abundance of a species in macro-ecology,or OTU abundance in diversity analysis. More detailed information on the dataset can be found in Doyle et al.(2018).
The Hill numbers(Hill,1973)were reintroduced to ecology by Jost(2007)and Chao et al.(2012,2014),and possess certain critical advantages over traditional diversity indexes.The Hill numbers for measuring alpha diversity are as follows:
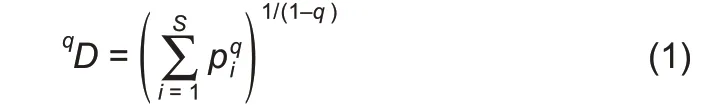
When q=1,the Hill number is undefined,but its limit exists in the following form:
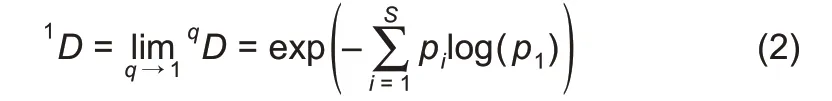
where,D is the diversity in Hill numbers,q(=0,1,2,…)is the order number of diversity,S is the number of species(or OTUs),and piis the relative abundance of species i.The diversity order(q)sets the sensitivity of the Hill numbers to the relative frequencies of species abundances.When q=0,0D is equal to the number of species or species richness(S).When q=1,1D is the number of typical or common species in the community and is equal to the exponential of Shannon entropy.When q=2,2D is more sensitive to species with high abundance,and is equal to the inverse of the Simpson index.Generally,qD is the diversity of a community with x=qD equally abundant species.
Beta-diversity can be defined with the multiplicative partitioning of Hill numbers(Chao et al.,2012,2014;Ellison,2010;Gotelli&Chao,2013;Jost,2007),as follows:

where,qDαandqDγare the alpha and gamma diversities in terms of Hill numbers,respectively.AsqDγis equivalent to the alpha diversity of the meta-community, it has the same definition as alpha-diversity (Eqn. (1)). Chao et al. (2012,2014) defined a series of Hill numbers corresponding to different diversity orders(q)as the diversity profile.In this study,the diversity or Hill numbers were computed until the third order,q=3.
According to Ma(2018a),we used the power law(PL)DAR model and power-law with exponential cutoff(PLEC)model as the DAR models for the human vaginal microbiome.The PL model is:

where,qD is diversity measured in Hill numbers of the q-th order,A is the area(number of individuals),and c&z are the PL parameters.
The PLEC model is:

where,d is a third parameter that is usually less than zero in DAR modeling,and exp(dA)is then the exponential decay item that eventually overwhelms the power law behavior when A is sufficiently large.
To simplify parameter estimation,we transformed non-linear Equations(4)and(5)into log-linear regression equations:
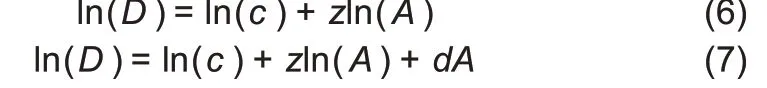
In Eqn.(6),z is the slope of the log-linear transformed PL model,which is equivalent to its interpretation in the traditional SAR—ratio of diversity accrual rate to area increase rate.Parameter c of the PL model can be viewed as the number of species equivalent to diversity in the first unit of area to accrue.Thus,the accrual order of area unit may influence parameter c. To deal with this technical issue, the units(individuals/samples) to be accumulated were randomly permutated each time the DAR model was built.For each dataset,we repeatedly applied DAR modeling 100 times by randomly re-ordering all samples in the dataset. For the detailed computational procedure,please refer to Ma(2018a).
Similar to the diversity profile concept of Chao et al.(2012,2014),which is a series of Hill numbers corresponding to different diversity orders(q),Ma(2018a)and Ma&Li(2018)proposed four DAR-based profiles,including the DAR,PDO,MAD, and LRD/LGD profiles. These four profiles can be quantitatively characterized by parameters from the PL/PLEC DAR models and can be used to sketch out biogeography maps of the human microbiome or other ecological communities.
The DAR profile was defined as a series of z-values(scaling parameter)of the PL-DAR model(Eqns.4&6),i.e.,a series of z-values corresponding to different diversity orders(q)or z-q trends.
The PDO profile was defined as:

where,z is the scaling parameter of the PL-DAR model,i.e.,the PDO profile is a series of g(q)values corresponding to different diversity orders(q),computed with Eqn.(8).
The MAD profile was defined as a series of MAD orqDmaxvalues,corresponding to different diversity orders(q):
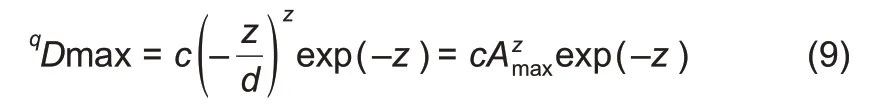
where, Amax=-z/d is the number of individuals (samples)needed to reach the MAD,and c and z are parameters of the PLEC-DAR model(Eqns.(5)&(7)).
The RIP profile was defined as a series of RIP values corresponding to different diversity orders(q),as specified by the following equation:

where,c is a parameter of the PL-DAR model and D is the diversity in Hill numbers estimated with the PLEC-DAR model(Eqns.(5)&(7)).Based on the above RIP definition,a RIP profile can be defined for a population(cohort)of any size.In practice,usingqDmaxforqD is more convenient,i.e.:

The RIP parameter assesses the average level of an individual to represent a population(or cohort)from which the individual is a member.The RIP profile is also known as the LRD (local-to-regional diversity) or LGD (local-to-global diversity) profile in other ecological systems beyond the human microbiome(Ma&Li,2018;Ma,2019).
We built two DAR models for the vaginal microbiome,including the PL and PLEC models for alpha-diversity and beta-diversity scaling,respectively.The results are listed in Tables 1 and 2, including the diversity order (q) of Hill numbers,mean model parameters(z,ln(c),d,g,Dmax)and their standard errors,and measures(correlation coefficient R&P-value)for goodness-of-fitting.N represented the number of successful fittings out of 100 p re-samplings,as explained previously. Re-sampling was performed to deal with the possible influence of the order of diversity accrual(i.e.,order in which the samples were accrued for building the DAR model)on model parameter c.Except for two cases of alpha-DAR modeling at diversity order q=3,the fittings to the DAR models were successful in all 100 re-samplings.Even in the two exceptions, the success rates were 97% and 99%,respectively. Therefore, the DAR models were considered suitable for vaginal microbiome assessment,as also evident by the R (linear correlation coefficient) and associated p values,which indicated the goodness-of-fit of the DAR models.
Based on Table 1,we found the following in regard to alpha-DAR scaling:
(1)As one of the most important parameters from the PLDAR model,the scaling parameter(z)at different diversity orders(q)was z(q)=(0.807(0),0.171(1),0.110(2),0.095(3)),where z(q)represents the DAR profile according to previous definition.The DAR profile characterizes the diversity scaling across individuals(over space)comprehensively.Results also showed that the scaling level differed at different orders.Forexample,scaling at diversity order q=0,which is equivalent to the classic SAR law,was faster than that at q=1,2,or 3,as indicated by the monotonically decreasing z-value(see Figure 1A for alpha-DAR profile).
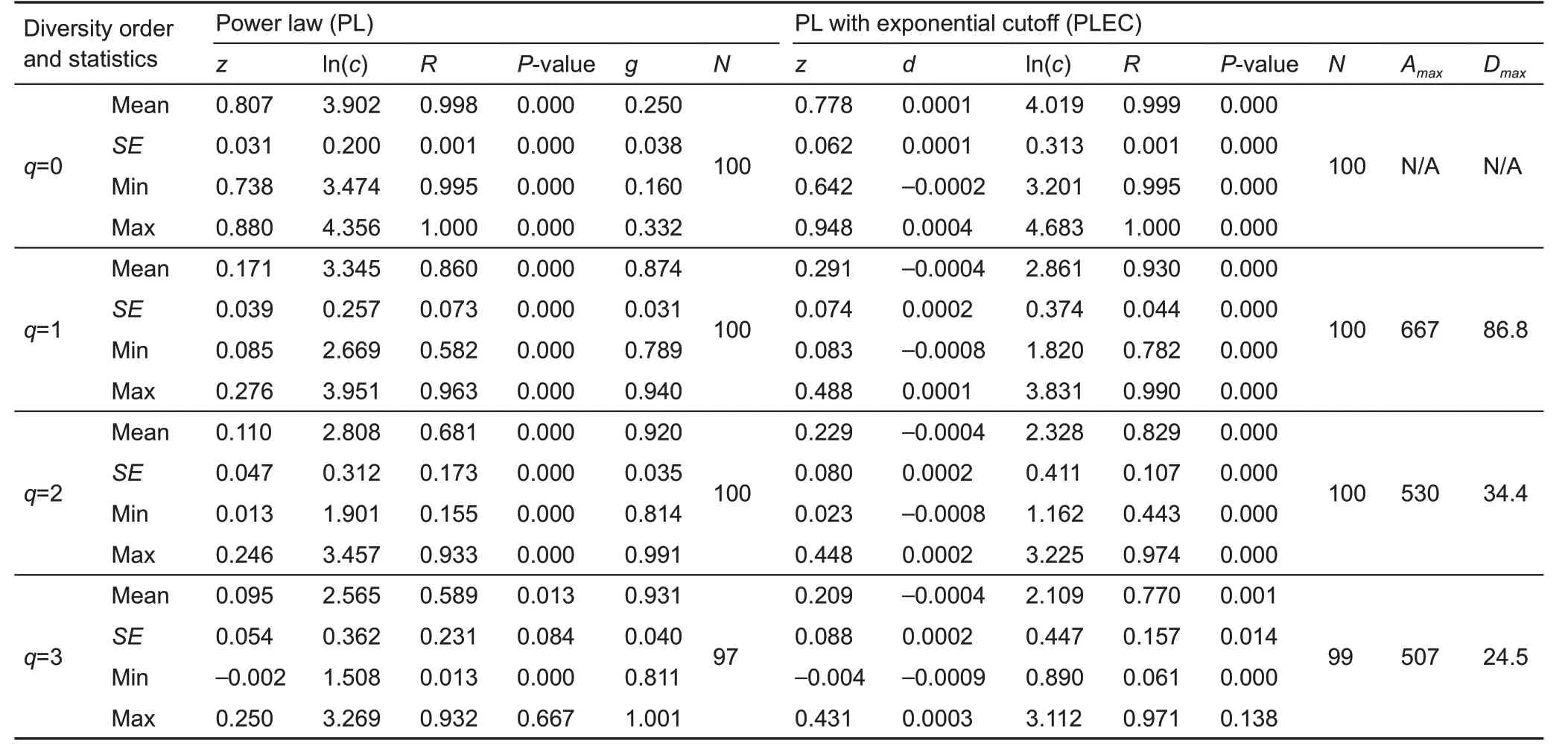
Table 1 Alpha-DAR models computed with 100 re-samplings for the vaginal microbiome
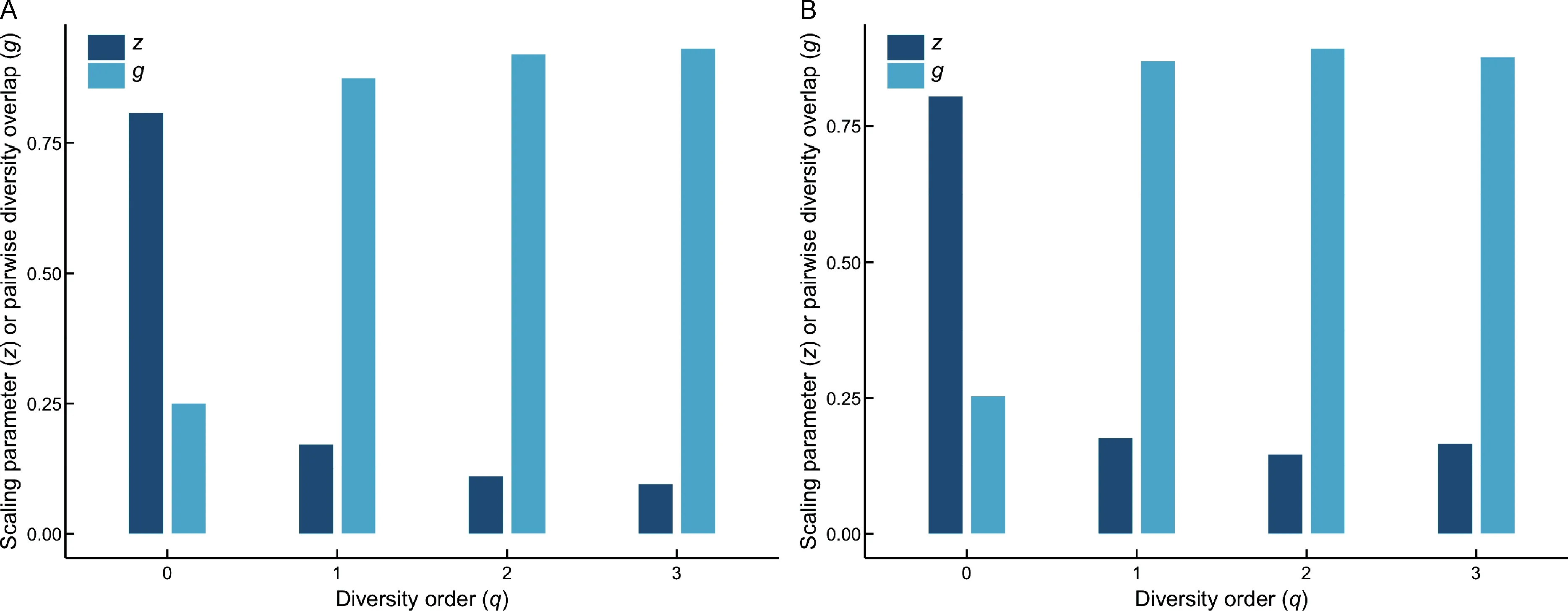
Figure 1 DAR profile(z-q)and PDO profile(g-q)of the vaginal microbiome
(2)The PDO profile was g(q)=(0.250(0),0.874(1),0.920(2),0.931(3)).The PDO profile,which characterizes the overlap or similarity between pair-wise individuals,showed the opposite trend as the DAR profile,i.e.,a monotonically increasing trend(see Figure 1A for alpha-PDO profile).
(3)The MAD profile characterizes the theoretically maximal accumulation of diversity across individuals.Here,regarding the MAD profile,the PLEC model failed to produce Dmaxat diversity order q=0 because d>0,for which a maximum does not exist.For the diversity orders q=1,2,3,the PLEC model for alpha-diversity successfully generated Dmax,i.e.,Dmax(q)=(86.8(1),34.4(2),24.5(3)).

Table 2 Beta-DAR models computed with 100 re-samplings for the vaginal microbiome
From Table 2,we found the following in regard to beta-DAR scaling:
(1)As one of the most important parameters of the PL-DAR model, the beta-diversity scaling parameter (z) at different diversity orders(q)was z(q)=(0.805(0),0.176(1),0.146(2),0.166(3)),where z(q)represents the DAR profile according to previous definition and characterizes diversity scaling across individuals(over space)comprehensively(see Figure 1B for beta-DAR profile).Comparison between the beta-DAR and alpha-DAR profiles revealed an interesting phenomenon:i.e.,the alpha-DAR profile monotonically decreased with q,whereas the beta-DAR profile was valley-shaped. This suggests that,at a lower diversity order(q),the alpha-DAR and beta-DAR scaling parameters(z)were rather close to each other,but the difference was enlarged at higher diversity orders(q).
(2)The beta-PDO profile was g(q)=(0.253(0), 0.870(1),0.893(2),0.877(3))for beta-diversity scaling.Here,the PDO profile,which characterizes the overlap or similarity between pair-wise individuals,showed the opposite trend to the DAR profile,i.e.,a bell-shaped trend(see Figure 1B for beta-PDO profile).
(3)The MAD profile characterizes the theoretical maximal accumulation of diversity across individuals.Here,regarding the beta-MAD profile,the PLEC model failed to produce Dmaxat diversity order q=0 because d>0,for which a maximum does not exist.For diversity orders q=1,2,3,the PLEC model for beta-diversity successfully generated Dmax,i.e.,Dmax(q)=(15.5(1),17.3(2),21.3(3)).
Table 3 shows the RIP values for both alpha-DAR and beta-DAR of the vaginal microbiome.At diversity order q=0,the estimation of0Dmaxfailed, and RIP for q=0 could not be estimated.For q=1,2,3,RIP was successfully estimated for alpha- and beta-diversity, respectively. Here, RIP characterized the relationship between individual- and population-level diversity.For example,at diversity order q=1,alpha-RIP=0.327 and beta-RIP=0.316, indicating that an average individual represented approximately 33%and 32%of population alpha-and beta-diversity,respectively.
In the current study,we investigated the diversity(including alpha- and beta-diversity) scaling of the human vaginal microbiome across individuals by re-analyzing a big datasetoriginally published by Doyle et al.(2018).Compared with the microbial SAR range reported in existing literature for other microbes, such as Green & Bohannan’s (2006) range between 0.019-0.470,the scaling parameter(z)estimated in our study,i.e.,alpha-z=0.807,beta-z=0.805,appears to be out of the known range,at nearly twice that reported for SAR values for other microbes.Three possibilities exist for the significant difference: (1) The use of revolutionary metagenomic sequencing technology, which allows for detection of more microbial species and consequently large scaling parameter;(2)The human vaginal microbiome has higher heterogeneity across individuals, which could be validated by future biomedical studies; and (3) The postpartum nature of the vaginal microbiome samples analyzed in this study. We could not exclude these possibilities at present due to insufficient available data for comparative research. Indeed, previous studies have classified human vaginal microbiomes into five main community-state types(CSTs),in which CST I,II,III,and V are dominated by Lactobacillus spp.,and CST IV is composed of facultative or strictly anaerobic bacteria(Gajer et al.,2012;Ravel et al.,2011),many of which are BV-related.The vaginal microbial communities of postpartum women in rural Malawi studied by Doyle et al. (2018) and reanalyzed here were mostly Lactobacillus-deficient microbiomes, which could be grouped as CST IV,although all these women were healthy.Therefore,the classification of CSTs may be more complex than initially conceived. Consequently, our DAR analysis based on Doyle et al.(2018)may be limited by the datasets of postpartum women,and the DAR parameters of the vaginal microbiomes of other CST women are likely different from the results reported here.Further studies should be performed to clarify this important issue.
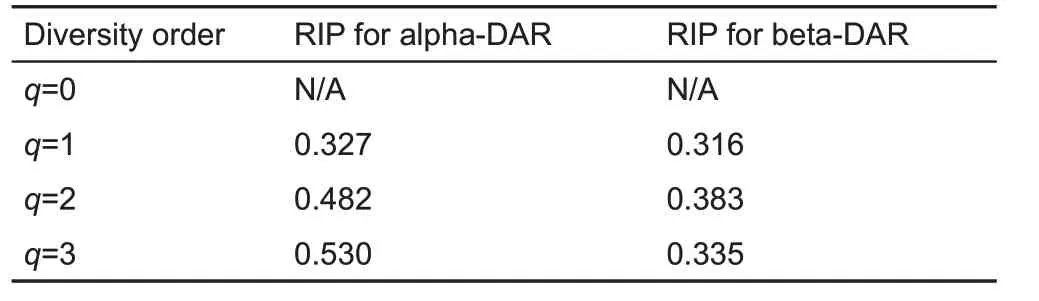
Table 3 Ratio of individual-level to population-level diversity(RIP)of the vaginal microbiome
The major findings in this study can be summarized using four profiles: i.e., DAR profile, characterizing the change(scaling)in diversity heterogeneity across individuals;PDO profile, characterizing the pair-wise similarity (overlap)between individuals;MAD profile,characterizing the maximal accrual diversity in a population; and RIP profile,characterizing the ratio of individual-level diversity to population-level diversity.Theoretically,the four profiles can together summarize the essential characteristics of the spatial distribution of vaginal microbial diversity and offer effective tools to sketch out the biogeographic maps of the human vaginal microbiome. Practically, they are essentially quantitative metrics of diversity heterogeneity across individuals from different dimensions(diversity scaling,pairwise similarity in diversity,maximal accrual diversity,ratio of individual to population diversity). These multidimensional metrics could provide more comprehensive tools for understanding the implications of vaginal microbial diversity to women’s health,including the DSR hypothesis for BV etiology(Ma et al.,2012,Ma&Ellison 2018,2019 Sobel,1999).In addition,the quantitative models of the four profiles obtained here could be harnessed to assess and predict microbiome diversity changes at the population scale and are of potential significance for evaluating women’s health associated with vaginal microbiomes.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS'CONTRIBUTIONS
Z.S.M.designed the study and wrote the paper.W.L.performed the data analysis and interpretation.All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We are deeply indebted to Prof.Yong-Gang Yao for his advice and review of our manuscript.We are also deeply grateful to Dr.Ronan Doyle,Great Ormond Street Hospital,NHS Foundation Trust,United Kingdom,for his assistance in re-analyzing the raw sequencing reads for this study.

- Zoological Research的其它文章
- Direct sunlight exposure reduces hair cortisol levels in rhesus monkeys(Macaca mulatta)
- Ongoing green peafowl protection in China
- First record of the ferret-badger Melogale cucphuongensis Nadler et al.,2011(Carnivora:Mustelidae),with description of a new subspecies,in southeastern China
- A new species of the genus Xenophrys(Anura:Megophryidae)from northern Thailand
- A new species of the genus Raorchestes(Anura:Rhacophoridae)from Yunnan Province,China
- Sinocyclocheilus sanxiaensis,a new blind fish from the Three Gorges of Yangtze River provides insights into speciation of Chinese cavefish