
偶氮苯光致异构储能材料的研究进展
2019-10-19 08:36:26江艳,黄金,罗文
广东工业大学学报 2019年5期
江 艳,黄 金,罗 文
(1. 广东工业大学 材料与能源学院,广东 广州 510006;2. 肇庆学院 机械与汽车工程学院,广东 肇庆 526061)
随着全球经济的高速发展,化石燃料已无法满足未来急剧增长的能源需求,同时增加了改变环境和气候的风险. 因此,发展高效的可再生能源技术已成为社会可持续发展的主要挑战[1-3]. 其中太阳能清洁无污染,取之不尽用之不竭,得到广泛关注,然而其是一种低密度、间歇性与空间分布不断变化的能源[4]. 因此,为了实现持续和高效利用太阳能,研究开发出将太阳能吸收、储存和转化成其他形式的能量以便于后用的有效技术显得尤为重要[5-6].
太阳能储能主要分为物理储能(显热储能和潜热储能)和化学储能(热化学储能和光化学储能)[7-8].在这些技术中,光化学储能是一种高能量、稳定的太阳能储能技术. 与热化学储能相比,光化学储能不需要对物质进行物理分离,也不需要将物质储存在不同的容器中,因此光化学储能尤其具有吸引力. 不同的光化学储能系统主要有降冰片二烯[9]、蒽[10]、二苯乙烯[11]、二钌富瓦烯[12]和偶氮苯[13]等. 由于偶氮苯化合物光致异构化的储能技术,可以实现高效便捷、稳定可控的热能储存/释放,近年来得到国内外的广泛研究[13-21]. 本文综述了偶氮苯光致异构储能材料储能机理、主要性能指标、偶氮苯化合物和模板化偶氮苯化合物的研究进展.
1 光致异构储能机理

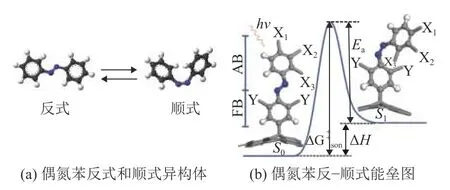
图 1 (a)偶氮苯反式和顺式异构体 (b)偶氮苯反−顺式能垒图Fig.1 (a) Trans and cis isomers of azobenzene (b) Energy barrier diagram of azobenzene trans-cis transition
2 性能指标
2.1 回复半衰期
偶氮苯光致异构储能材料的回复半衰期是其一个重要性能指标. 偶氮苯化合物将光能储存在顺式亚稳态的化学键中,该亚稳态结构即使不受外部条件激发也会自发向反式稳态转变. 在常温下,大多数偶氮苯化合物的回复半衰期一般只有几秒~几分钟[33-35],使得偶氮苯化合物的能量不能长时间储存,因此提高回复半衰期能使偶氮苯化合物用于长期储能.
2.2 能量密度
能量密度是储能材料又一主要指标,其体现储能材料的储能能力,由顺式和反式之间的能级差决定[28].
相比于其他储能材料或储存设备,虽然偶氮苯储能材料有简单可循环储能和较低成本的优点,但是传统的偶氮苯化合物还是存在着储能密度低的问题. 因此如何在分子结构设计和模板化结构下,进一步提高偶氮苯储能材料的能量密度是该类储能材料研究领域的又一研究重点.
2.3 储能范围
传统的偶氮苯化合物只能通过紫外光照射进行反−顺式异构化,而太阳光谱中紫外光的含量只有4.5%,严重阻碍了偶氮苯储能材料的效率. 因此开发拓宽偶氮苯化合物的储能范围能有效提高太阳光的利用效率.
为了能够解决传统偶氮苯分子普遍存在储能密度低和/或稳定性差(回复半衰期短)和/或需要紫外光储能的问题[10],一个可能的解决方案是在分子设计中使用推−拉型电子结构、强给电子基团[36-37]、多邻位取代基[38-40]或多偶氮苯[41-42];另一种方法是设计含有软聚合物或刚性碳纳米材料模板化结构,通过增强分子间相互作用和空间位阻来增加能量密度[12]和/或半衰期或可见光储能. 以下就从偶氮苯化合物和模板化偶氮苯化合物的研究进展进行综述.
3 偶氮苯化合物的研究现状
英国科学家Hartley于1937年首先发现顺式偶氮苯[43]. 随后测量出偶氮苯的储存焓(顺反式异构体之间的能量差)[44-47]. 为了探究偶氮苯化合物储存太阳能的潜力,科学家用不同极性取代基对偶氮苯双苯环进行取代,结果发现,取代基能降低顺式偶氮苯的热回复寿命、光吸收效率和能量密度以及在极性溶剂中有限的溶解度[47-48]. 然而,提高偶氮苯化合物半衰期或能量密度仍然是关键.
在提高偶氮苯化合物顺式异构体回复半衰期研究中,Woolley及其同事[49]调整了一系列对位取代偶氮苯中取代基的相对给电子能力,不仅调节了偶氮苯的吸收光谱,发现其π-π*带的波长从sp3碳(342 nm,1)、酰胺(366 nm,2)、氨基甲酸酯(372 nm,3)至脲取代基(382 nm,4)(见图2),而且极大地影响了顺式异构体的半衰期,sp3碳偶氮苯衍生物(见图2中1)的半衰期为43 h,富电子脲偶氮苯衍生物(见图2中4)的半衰期为11 s,表明偶氮苯的π-π*带红移越多表现出更快的热弛豫过程[50-52];Saalfrank等[53]运用量子化学计算方法系统研究了在不同溶剂中不同类型和数量的取代基和其在苯环上不同位置对偶氮苯化合物热回复顺−反异构化速率的影响. 研究表明具有邻位给电子取代基(-OMe,-NH2)的偶氮苯衍生物比纯偶氮苯分子更可能具有更高的能垒(Ea),即半衰期更长. 这种大的Ea与紧凑顺式异构体的空间不稳定性有关[54]. 同时该类偶氮苯的热异构化速率很大程度取决于溶剂,相对于非极性溶剂,在极性溶剂中其速率可提高2个数量级. 这是由于在极性环境中异构化具有较低障碍.
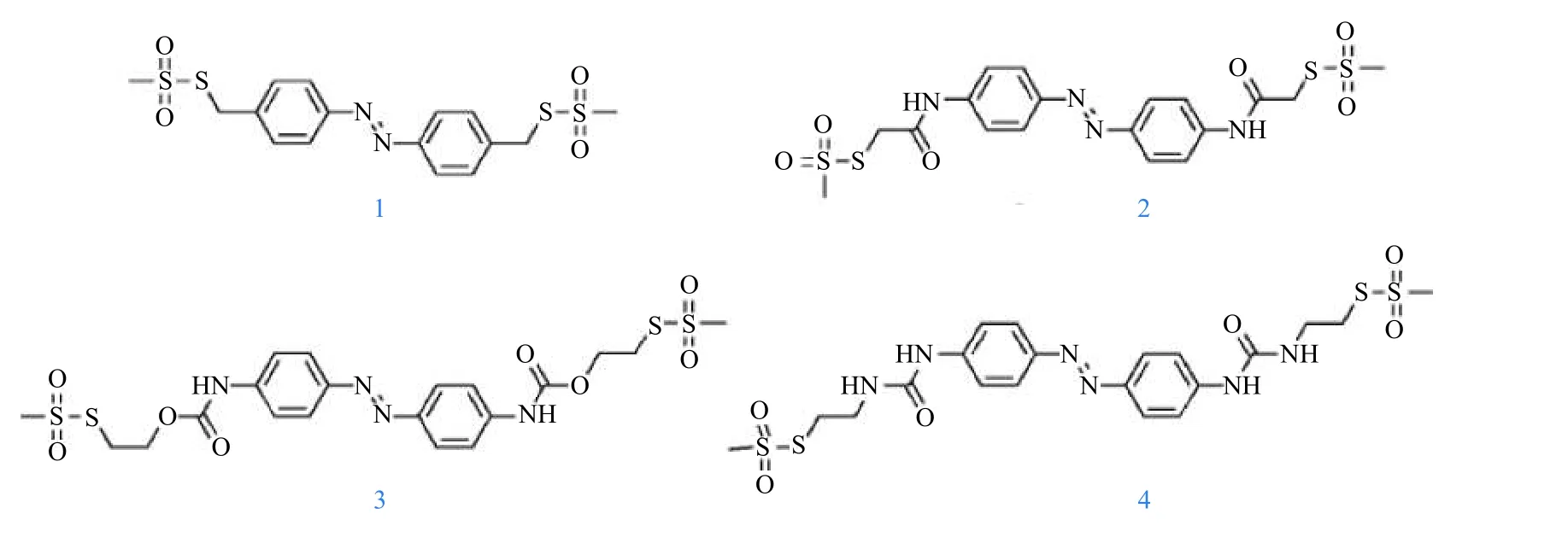
图 2 甲烷硫代磺酸盐(MTS)烷基-AZO(1)、MTS-酰胺-AZO(2)、MTS-氨基甲酸酯-AZO(3)和MTS-脲-AZO(4)的结构[49]Fig.2 Structure of methane thiosulfonate (MTS) alkyl-AZO (1), MTS-amide-AZO (2), MTS-carbamate-AZO (3) and MTS-urea-AZO (4) [49]
Herges和Yan等[55-56]制备和研究了2,2′-乙烯桥联偶氮苯(br-Azo),研究发现引入乙烯桥,其顺式和反式异构体的n-π*带出现明显的分离,其中的一些衍生物具有相对较长的半衰期,由于存在环张力,顺式异构体为热力学稳定形式. 随后,Herges课题组[57]成功合成含氧和硫桥的偶氮苯衍生物,研究发现这两种偶氮苯衍生物能够用650 nm的远红外光进行异构化,相比于之前的乙烯桥偶氮苯具有更长的半衰期,达到2.5 d. Wolley等[35]用给电子无环或环状氨基取代基取代偶氮苯的2,2′-位,制备了一系列具有不同立体和电子效应的偶氮苯衍生物,六元环状取代基((5)或(6))与偶氮苯环具有空间相互作用,导致环状取代基的扭曲和N-原子上的sp2特征的丧失. 五元环(8)或无环(7),可以减轻空间碰撞,并且电子给予的程度可以最大化. 特别有趣的是(6)能吸收蓝光并保留与其对位取代相当的转换波长,但其具有~600倍慢的热弛豫率(见图3),说明相对于小环或无环,大环取代基能增加空间阻碍,延长该类取代基偶氮苯的顺−反式异构化寿命. Grossman等[58-59]设计了新型的光控热能储存材料,该类材料由偶氮苯分子和有机相变材料组成,通过将对位改性为具有和相变材料具有类似碳链的偶氮苯分子,提高了偶氮苯分子与相变材料分子间的相互作用,从而提高了偶氮苯顺式异构体的半衰期,达到10 h以上.
Hecht研究小组[41]制备4种线性结构双偶氮苯衍生物. 研究表明全顺式异构体(Z,Z)回复到全反式异构体(E,E)需要经历两个步骤,中间还需经历(Z,E)过程,即逐个偶氮键回复. 室温条件下,由于环张力或π-堆叠趋势(几何或电子耦合),其回复半衰期从10至57 h. Sönnichsen等[60]由修饰Ullmann反应合成了双-和三-偶氮苯(BPAPA和TPAPA),在室温下以稳定的全反式(E)存在. 实验结果表明在乙腈溶液中35 ℃条件下,顺式(Z)BPAPA热回复反应的半衰期为0.5 h(Z,E)和2.4 h(Z,Z). 顺式TPAPA热回复反应的半衰期为4.0 h(Z,E,E)、6.4 h(Z,E,E)和12 h(E,E,E). 结果表明顺式异构体的半衰期随着生色团的尺寸和偶氮苯单元数量的增加而显著增加[41],这种行为是由于分子间的相互作用和双/三偶氮的分子内相互作用导致了同分异构体能量和几何结构的剧烈变化.

图 3 2,2′-位含氨基取代基的4,4′-二乙酰氨基偶氮苯的结构式[35]Fig.3 Structures of 4,4′-diacetylaminoazobenzenes containing an amino substituent at the 2,2′-position [35]
Nishioka团队[61-62]报道了2′,6′-二邻位取代的方法,引入给电子甲基,利用氮原子π轨道孤对电子和远端苯环上甲基之间的空间位阻导致反转过程延迟,实现了在60 ℃下半衰期超过25 h,而且通过改变溶剂极性,该类偶氮苯衍生物在不同极性溶剂中会产生溶剂效应. 随着溶剂极性下降,该偶氮苯的半衰期呈现下降趋势,即溶剂极性下降,半衰期变短. 这种溶剂效应表明其顺式异构体的热异构化是通过反转机制进行而不是旋转机制;Han等[63]设计并制备出邻位乙基(空间庞大)双取代偶氮苯Et-SH,经365 nm紫外光照射至光稳态后,在室温下热回复顺−反异构化速率非常缓慢,半衰期高达380 h,是相同结构间位甲基单取代偶氮苯(13 h)的30倍. Et-SH如此慢的异构化速率归因于在异构化过程中空间庞大的乙基取代基抑制了偶氮基团的大规模变形.
综上所述,发现在偶氮苯的邻位进行增加空间相互作用的取代能有效提高偶氮苯衍生物的半衰期,但是所有取代的偶氮苯衍生物只能通过紫外光激发反−顺异构化,因此开发半衰期长和能可见光激发反−顺异构化的偶氮苯衍生物是非常必要.
在用可见光激发偶氮苯化合物反→顺异构化的研究中,2,2′-偶氮苯萘可以分别用蓝光和绿光来诱导E→Z和Z→E异构化[64],然而扩展π系统能有效红移通常伴随半衰期的显著降低(从几天到几小时或更小). 降低该能隙并因此使偶氮苯吸收红移的一种经典方法是在N=N的邻位或对位引入给电子基团(EDGs)和/或吸电子基团(EWGs)(氨基偶氮苯或推−拉偶氮苯),因此π-π*带红移到400~600 nm区域,但Z异构体的热稳定性降低,这两种效应随供体/受体取代基的强度而增强[65-69].
Woolley等[38]采用氧化偶合法制备了对称酰胺基偶氮苯分子,研究发现在25 ℃二甲基亚砜中,所有4个邻位被甲氧基取代的偶氮苯的顺、反式两种异构体的n-π*带出现大的分离(顺式异构体的n-π*跃迁相比于反式异构体的蓝移36 nm),并且顺式异构体的半衰期为53 h. 经过含时密度泛函计算得到相关验证,并解释出现这种大分离是因为比较映射到键密度表面上的每种异构体的最高占据分子轨道(HOMO)的绝对值(见图4). 基于相同的原理,Woolley等[70-71]报道了所有四邻位被富电子大取代基(甲氧基、氯和溴)取代的偶氮苯化合物,这类偶氮苯分子的顺式异构体也具有非常长的半衰期. Hecht等[40,72]报道了一类新型的四邻位氟取代偶氮苯,他们发现邻位氟取代基的存在为传统的偶氮苯引入了两个新特征. 首先,在发色团的反式和顺式异构体的可见光区域(nπ*)出现明显的光谱分离. 这种分离是由氟原子的吸电子能力降低了N=N键附近的电子密度,从而降低n-轨道的能量. 使得反式异构体吸收最小的区域(>500 nm),而顺式异构体吸收最大的区域(约410~420 nm). 其次,也许更重要的是,25 ℃下乙腈溶液中顺式异构体表现出惊人的热稳定性,其半衰期约高达2年.

图 4 (a)对称酰胺基偶氮苯分子,(b)和(c)分别为计算的反式和顺式结构. 每种异构体HOMO的绝对值映射到键密度表面(蓝色是HOMO的最大值)[38]Fig.4 (a) Symmetric amido azobenzene molecules, (b) and (c)calculated structures of trans- and cis-a. The absolute value of the HOMO of each isomer is mapped onto the bond density surface (blue is the maximum value of the HOMO) [38]
2012年,Aprahamian和共同作者[73]报道了一类新型的偶氮苯,研究发现BF2与偶氮基团的氮孤对子的配位导致n-π*和π-π*跃迁能级的位置反转,这种现象导致两种异构体具有明显的πnb-π*分离带,用紫外−可见吸收光谱监测其顺−反式异构化,发现在20 ℃其半衰期为12.5 h. 而且密度泛函理论计算表明电子密度的增加可能会使系统中的吸收带红移,并且将给电子基团接枝到BF2-偶氮苯的对位和邻位. 实验证明验证了这种现象[73-74]. Bai等[75]系统研究了在其对位引入给电子基团BF2-偶氮苯的热顺−反式异构化,结果发现不同的电子给体基团可以显著影响吸收光谱、分子轨道的能级、反式异构体的过渡性质、速率常数和顺−反式异构化的半衰期,随着给电子能力的增强,顺−反式异构化速率增加,即对应半衰期相应减小.
由此可见,先前大部分研究都集中在提高偶氮苯衍生物的半衰期上,而其作为一种储能材料,研究并提高偶氮苯化合物的能量密度成为关键.
在提高偶氮苯分子的能量密度(摩尔或质量能量密度)研究中,19世纪80年代,Adamson等[46]利用光量热法测量出偶氮苯的储存焓为49 kJ·mol−1. 封伟等[76-78]制备邻位取代偶氮苯,研究发现其能量密度为72 kJ·mol−1(40 W·h·kg−1). 随后,该课题组[42,79-80]基于多偶氮能提高储能密度[41,60]合成了双和三偶氮苯分子(bis-AZO和tri-AZO),由于bis/tri-AZO的分子内相互作用导致异构体的能量和几何结构发生显著变化,结果显示bis-AZO和tri-AZO的储存能量分别为80.9 kJ·mol−1(32 W·h·kg−1)和135.1 kJ·mol−1(40.7 W·h·kg−1).Kimizuka等[81]展示了一种液态偶氮苯衍生物,通过对不同异构化程度的该偶氮苯进行DSC测试及其分析,计算得到100%异构化的异构化焓为52 kJ·mol−1(47 W·h·kg−1). CHO等[82]设计并制备了一类将庞大芳基(苯基、联苯和叔丁基苯基)接枝到偶氮苯上的衍生物(见图5中9~11),由于排斥和空间位阻,引入芳基能使顺式偶氮苯的能态升高,从而增加反式和顺式异构体之间的能量差,热量释放的能量密度达到87 kJ·mol−1(108 J·g−1或30 W·h·kg−1),制备得到的分子固态膜,由于存在所需的自由体积能够光诱发反−顺异构化,而且通过分子设计,充电率提高了60%,充电能力提高了33%. Grossman研究组[83]报道了一系列偶氮苯功能化的丁二炔(见图6),研究发现每摩尔功能化丁二炔反向异构化的能量(ΔH)随着连接偶氮苯间隔烷基链的增长而增加,从6-a的121.9 kJ·mol−1(209.9 J·g−1)到6-c的 160.1 kJ·mol−1(222.1 J·g−1),与原始偶氮苯相比分别增加47%和93%.此外,发现ΔH与连接部分的基团有关,带有酯基最长烷基间隔基的化合物6-d在该系列中释放出最大的能量176.2 kJ·mol−1(243.7 J·g−1),与带有酰胺基的图6-c和原始偶氮苯相比,其ΔH分别增加10%和113%. 随后,Grossman等[58-59]设计制备出了对位与有机相变材料(PCM)具有类似碳链结构的长链偶氮苯(见图7中12),差式扫描量热法测试其复合材料的能量密度为200 J·g−1(56 W·h·kg−1).
在上述偶氮苯分子结构设计中(主要性能比较如表1所示),通常在偶氮苯母体的邻或对位进行取代能较大影响其光化学特性,即对位引入推−拉型电子结构、对位强给电子基团和多邻位取代(甲氧基或卤原子),研究均表明,仅从分子结构本身的设计角度出发,得到同时具有高能量密度、长半衰期和可见光储能的偶氮苯衍生物是困难的,需通过引入其他结构的材料(碳纳米材料和聚合物)来调节储能化合物本身的物理性质.

图 5 (a)大体积苯基功能化偶氮苯衍生物的分子结构,(b)顺式−偶氮苯衍生物的DSC曲线[82]Fig.5 (a) Molecular structure of bulky phenyl group-functionalized azobenzene derivatives, (b) DSC curves of cis-azobenzene derivatives [82]
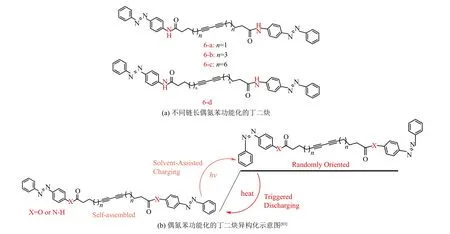
图 6 (a)不同链长偶氮苯功能化的丁二炔(b)偶氮苯功能化的丁二炔异构化示意图[83]Fig.6 (a) Diacetylenes functionalized by different chain length azobenzenes (b) Schematic diagram of diacetylenes isomerization [83]
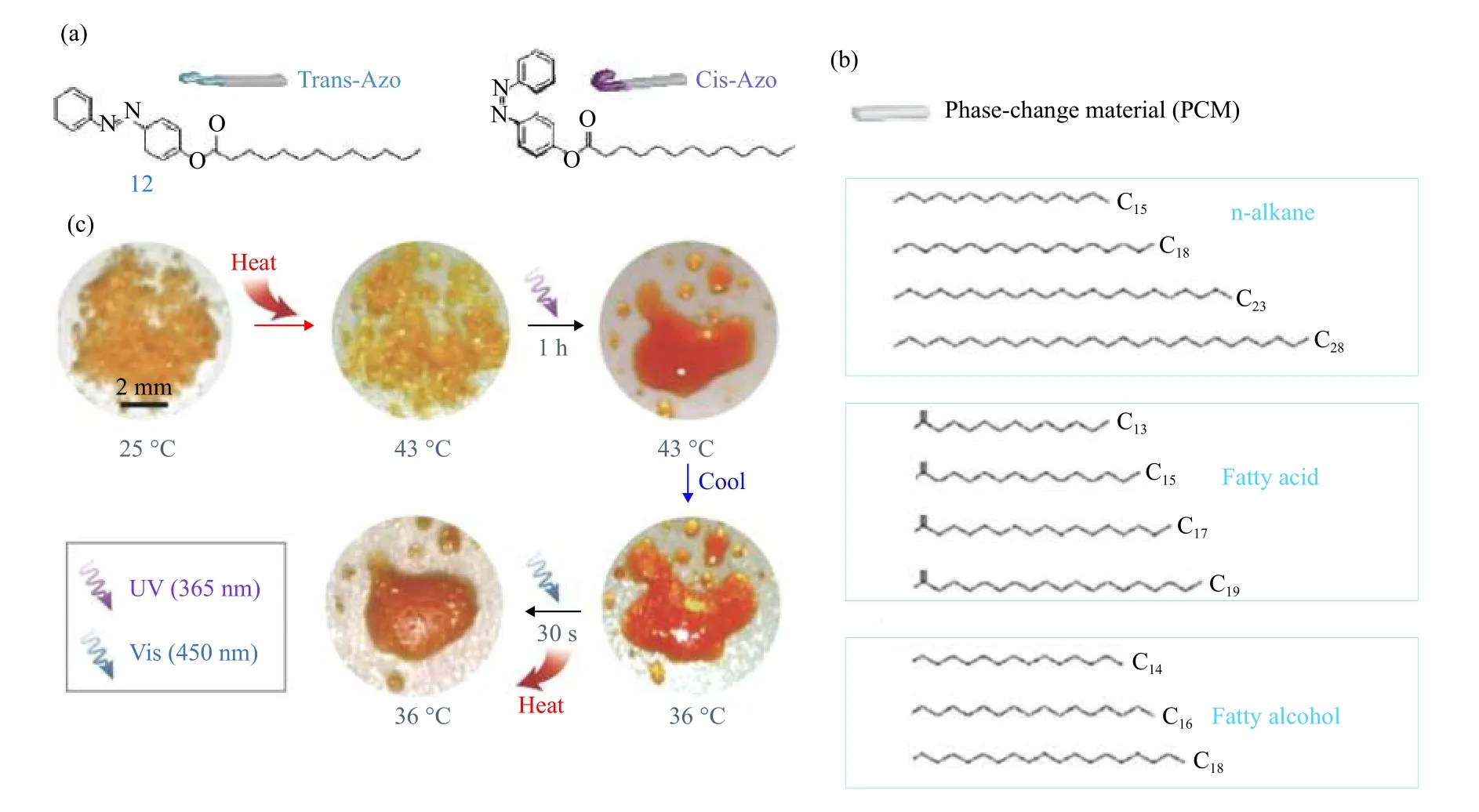
图 7 (a)长链偶氮苯的化学结构(b)多种n-烷烃、脂肪酸和脂肪醇相变材料的化学结构(c)相变材料和长链偶氮苯掺杂剂的复合材料的固体热吸收/紫外充电/冷却/可见光诱导放电和放热过程[58,59]Fig.7 Chemical structures of (a) long-chain Azo, (b) various n-alkanes, fatty acids and fatty alcohols, (c) Photographs of composites of PCM and long-chain azobenzene dopants during solid-state heat absorption/UV charging/cooling/visible-light-induced discharging and heat-release process [58,59]
4 模板化偶氮苯化合物的研究现状

图 8 (a)不同末端偶氮苯聚合物化学结构(b)从THF和DCM干燥的顺式(8c)的DSC曲线(c)基于不同THF和DCM比例混合溶剂中的(8c)聚合物的能量密度和FWHM图[84]Fig.8 (a) Chemical structure of different terminal azobenzene polymers (b) DSC curve of cis (8c) dried from THF and DCM, and (c) energy density and FWHM diagrams of (8c) polymer in mixed solvent based on different ratios of THF and DCM [84]
表 1 不同取代偶氮苯化合物的性能比较Tab.1 Comparison of properties of different substituted azobenzene compounds
在聚合物模板化偶氮苯化合物用于储能材料的研究中,Venkataraman等[84]开发了偶氮苯基间同立构聚(甲基丙烯酸酯)聚合物(图8(a)中8a-c),研究了处理溶剂和薄膜结构在实现更高能量密度方面的关键作用. 研究发现从二氯甲烷(DCM)干燥的样品的放热范围比从四氢呋喃(THF)获得的样品宽,但能量密度更低(图8(b)). 具体而言,从DCM干燥的样品的平均能量密度为110 J g−1(30.6 W·h·kg−1),而从THF获得的样品的平均能量密度为510 J·g−1(142 W·h·kg−1). 从THF和DCM混合溶剂中干燥,随着DCM比例的增加,能量密度呈现下降趋势(图8(c)). 主要原因是DCM优先与聚合物主链产生相互作用,而不会将顺式−偶氮中的偶极子溶剂化,导致在溶液中的聚集.这种聚集进一步导致紧密的聚合物间堆叠、偶极子的无序排列以及聚合物主链之间的体积不足以在异构化后的侧链顺式−偶氮之间进行π-π堆积. 最后,这些因素产生了缺乏协同异构化的固体活性层,从而导致较低的能量密度. 据推断,处理溶剂应充分溶剂化在充电状态下形成的偶极子,以减少溶液中的聚集,并产生允许有效π-π堆叠以实现高能量密度的最佳结构.
Morikawa等人[85]开发了一种偶氮苯衍生物的离子晶体(ICs),它由具有各种亚甲基长度(m)和烷基尾(n)的间隔基,低聚(环氧乙烷)基铵基团和反离子(X)组成(图9a,13). 聚合物模板化的偶氮苯IC在UV照射下可以发生从IC到离子液体(IL)的相变. 然后,IL通过可见光照射结晶成IC. 这种转化伴随着总放热焓为97.1 kJ·mol−1(128 J·g−1或36 W·h·kg−1),是纯偶氮苯异构化中储存能量的两倍多. 而且发现在34~65 ℃的温度范围内可以观察到两个共存的放热峰. 宽峰归因于偶氮苯的热顺−反式异构化,而46 ℃左右的急剧放热峰归因于IL向IC的相变焓. 随着继续加热,在87 ℃出现急剧的吸热峰,这与IC的熔化有关. Grossman研究组[83]报道了一系列偶氮苯功能化的丁二炔,在紫外光照射下,丁二炔单体在刚性共轭骨架上快速光聚合成含有偶氮苯侧链的聚丁二炔(图10,14),这种结构类似于装饰有紧密堆积的光响应单元的聚合物模板. 研究发现制备的材料通过偶氮苯的光异构化能发生相变(偶氮苯的反式构型自组装成晶体结构,顺式构型通过异构化过程变为液体),而且聚合物均能获得较高能量密度为173 kJ·mol−1(239 J·g−1或66 W·h·kg−1).

图 9 (a)阴离子偶氮苯衍生物化学结构(b)顺式-1(6,4)-Br的DSC曲线[85]Fig.9 (a) Chemical structure of anionic azobenzene derivative (b)DSC curve of cis-1(6,4)-Br with different structures [85]
Grossman[86]设计了一种简单的侧链聚合物,即主链为烷基链,侧链为偶氮苯(图11a,15),在室温下,该材料呈现相对较长的半衰期,聚合物的半衰期为55 h,能量密度分别为29 W·h·kg−1(104 J·g−1). 将此聚合物经过旋涂过程得到旋涂膜,研究此旋涂膜的热释放,结果发现与未充电膜相比,充电膜在几十秒内产生的热量在宏观上的表现为平均温差达到10 ℃,可用于某些快速放热应用中. Grossman研究组[87]设计了新型聚合物模板化偶氮苯,这种聚合物是将偶氮苯和羧酸基单体共聚合获得(图11c,16),电沉积到不同种类的基材上得到聚合物薄膜. 研究发现该聚合物膜能储存85~95 J·g−1(24~26 W·h·kg−1)的能量,并且处于顺式异构体状态的该薄膜的半衰期为75 h. 最近,德国马普所吴思等[88]报道了一种新的偶氮苯聚合物(PmAzo, 18),该PmAzo是以含有邻甲氧基取代偶氮苯(mAzo)基团的己基甲基丙烯酸酯为单体,并通过使用不同波长的可见光显示可逆的反−顺式或顺−反式异构化. 研究发现,该偶氮苯聚合物顺式异构体的半衰期为16 h,顺式和反式两种构型间的能量差为12.2 kJ·mol−1. 随后,吴思课题组[89]提出了一种能有效储存紫外光和可见光的偶氮苯聚合物(PAzo, 17;PmAzo, 18),该研究发现,PAzo和PmAzo两者储存的能量分别为54.8 kJ·mol−1(125.2 J·g−1或34.8 W·h kg−1)和12.2 kJ·mol−1(14.4 J·g−1或4.4 W·h·kg−1). 两者之间的差异是因为相比于PAzo,PmAzo邻位的甲氧基增加了反式构型的立体相互作用,而且PmAzo对较长波长的可见光也敏感. 两种聚合物在溶液中的热松弛顺−反异构化的半衰期分别为12.45 h和16 h.
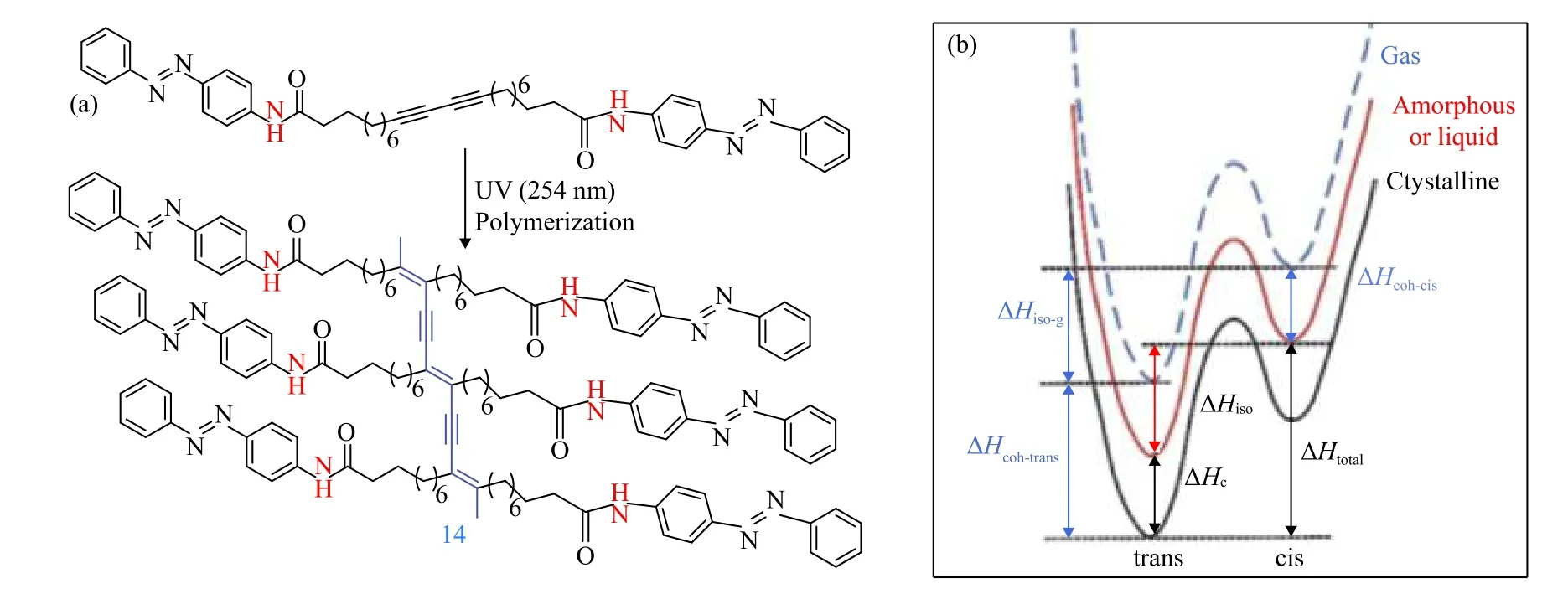
图 10 (a)光聚合生成聚合物结构的示意图(b)不同相态下偶氮苯衍生物的能量示意图[83]Fig.10 (a) Schematic diagram of photopolymerization to form a polymer structure (b) schematic diagram of energy of azobenzene derivatives in different phase states [83]
综上所述,聚合物模板化偶氮苯虽然能成膜易集成于元器件中,具有固态可控储能和热释放(主要性能比较如表2所示),但聚合物模板化偶氮苯化合物的能量密度低和半衰期短.
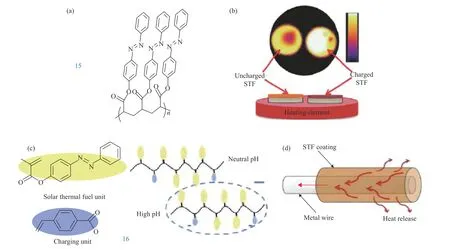
图 11 (a-b)偶氮聚合物的化学结构和其薄膜的热释放[86],(c-d)偶氮聚合物的化学结构和热释放[87]Fig.11 (a-b) Chemical structure of Azo-polymers and heat release in Azo-polymer films [86], (c-d) Chemical structure of Azo-polymers and heat release into the metal wire [87]

图 12 (a-b)偶氮聚合物的化学结构,(c)偶氮聚合物的DSC曲线[88, 89]Fig.12 (a-b) Chemical structure of Azo-polymer, (c) DSC curves of Azo-polymer [88, 89].
在刚性碳纳米模板化偶氮苯材料同时提高储能密度和半衰期的研究中,2011年,Kolpak和Grossman[90]提出引入刚性、低质量的碳纳米管(CNTs)作为模板,偶氮苯分子能在碳纳米管上形成高度有序且紧密排列的阵列. 在高接枝密度下,与单独的偶氮苯分子相比,CNT基质的存在可能破坏偶氮苯的分子对称性,形成紧密结晶状态,通过苯环的旋转防止异构化反应,从而增加形成氢键的潜在相空间,则处于纳米模板表面相邻的两个偶氮苯分子间距离明显缩小,导致分子间作用力显著增大,增强的分子间相互作用力改变了分子的能级,提高了其ΔH和Ea,从而提高其能量密度和回复半衰期. 随后,Grossman研究小组[91]使用第一性原理计算理论证明,将偶氮苯与不同碳基纳米模板(石墨烯、碳纳米管和富勒烯)结合,预测这些材料能可逆地储存太阳能,其能量密度可以和锂离子电池相媲美,具有高达35%的潜在外部效率,可以调节其热稳定性从几分钟到几年不等. 2014年,Grossman等[92]将CNTs模板化偶氮苯杂化材料从理论模型变为现实,他们采用多次有机自由基化反应制备高接枝密度(1:19)的偶氮苯-碳纳米管杂化材料(图13a,19). 与未模板化偶氮苯分子相比(58 kJ·mol−1或~160 J·g−1或~44 W·h·kg−1),在没有氢键调控的情况下,仅通过分子间相互作用和穿插效应(图13b)的影响,显示出每分子储存的能量的明显改善,其能量密度达到120 kJ·mol−1(~200 J·g−1或~56 W·h·kg−1),同时保持优异的循环性和稳定性. 随后,黄金等[37]报道了偶氮苯-多壁碳纳米管杂化材料(AZO-MCNTs),通过增强的分子间相互作用和穿插效应实现了高能量密度(77 W·h·kg−1)和较长半衰期(14 h),相比于偶氮苯分子本身的能量密度提高92.5%,半衰期提高了2个数量级. 最近,黄金等[93]通过将偶氮苯共价接枝到单壁碳纳米管上,通过分子内和分子间相互作用以及捆绑效应,相对于偶氮苯分子其能量密度提高107%(80.7 W·h·kg−1),半衰期为16 h.

表 2 不同聚合物模板化偶氮苯化合物的性能比较Tab.2 Comparison of properties of different polymer-templated azobenzene compounds

图 13 (a)碳纳米管模板化偶氮苯体系光化学储能和热能释放反应,(b)偶氮苯-碳纳米管杂化材料的穿插效应[92]Fig.13 (a) Reaction scheme for photo-chemical energy storage and thermal energy release in CNT-templated azobenzene systems, (b)Interpenetrating effect of azobenzene-carbon nanotube hybrid materials [92]
封伟等[94]运用计算机模拟技术构建了一系列石墨烯-模板化偶氮苯杂化物模型,其中在不同位置(邻位、间位和对位)具有各种官能团的光响应性偶氮苯与石墨烯共价结合. 密度泛函理论计算预测石墨烯上两个相邻偶氮苯分子之间的分子间距离、电子相互作用、空间位阻和分子间或分子内氢键导致ΔHstorage的变化. 封伟课题组[76]首次报道高接枝密度的邻位/对位取代的偶氮苯-石墨烯杂化材料通过氢键调控分子能量密度和半衰期,通过分子内/分子间氢键调控分别得到了长半衰期(5 400 h)和较高的能量密度(269.8 kJ·kg−1). 2015年,封伟课题组[77-78]报道了采用多次重氮盐自由基法合成了多种接枝密度高达1:16~1:19的邻位(间位或对位)含有甲氧基和/或羧基的偶氮苯-石墨烯杂化材料(图14,20). 在高接枝密度下,偶氮苯分子能够在石墨烯表面形成多重分子间氢键,提高了顺反异构体的能级差和顺式结构的半衰期,在保持了偶氮苯本身优异的光响应性和循环稳定性的基础上,通过氢键作用得到了高能量密度(112~138 W·h·kg−1)和较长的半衰期(792~1 250 h)定性的偶氮苯-石墨烯杂化材料. 理论上,双/三偶氮的分子内偶联不仅降低了反式异构体的能量,而且由于大的空间位阻而稳定了顺式异构体. 最近,封伟等[42,79]提出了模板组装的密堆积双偶氮苯共价接枝到还原氧化石墨烯上(RGO-bis-Azo,21-23),通过密度泛函理论计算的双偶氮(反式和顺式异构体)的空间构型和能量受到高接枝密度和捆绑效应引起的分子内和分子间空间位阻的影响. 这导致异构化的焓和活化能显著增加. RGO-bis-Azo杂化材料的高能量密度为80~131 W·h·kg−1,最大功率密度为2 230~2 517 W·kg−1,顺式异构体的回复半衰期为1 120~1 320 h,可调节的热释放时间为2 min至5 520 h. 对由该杂化材料形成的自支撑膜进行热释放研究,发现充完电后的薄膜与未充电薄膜之间的最大温差高达15 ℃,并且释放85%热量的持续时间为8 min. 2018年,封伟课题组[80]将三偶氮共价接枝到RGO上(RGO-tri-Azo, 图14,24),通过控制分子间相互作用,RGO-tri-Azo的能量密度高达150.3 W·h·kg−1,顺式异构体的回复半衰期为1 250 h,最大功率密度为3 036.9 W·kg−1.该类自组装膜在充放电过程中显示变色过程且释放的能量可以将膜的温度提高6~7 ℃.
表3总结了碳纳米模板化偶氮苯化合物的储能密度和回复半衰和反−顺异构化激发方式. 根据上述相关研究,通过提高偶氮苯在碳基纳米模板上的接枝密度以及分子间氢键和捆绑效应有效提高了偶氮苯/碳纳米模板化复合材料的半数期和能量密度.
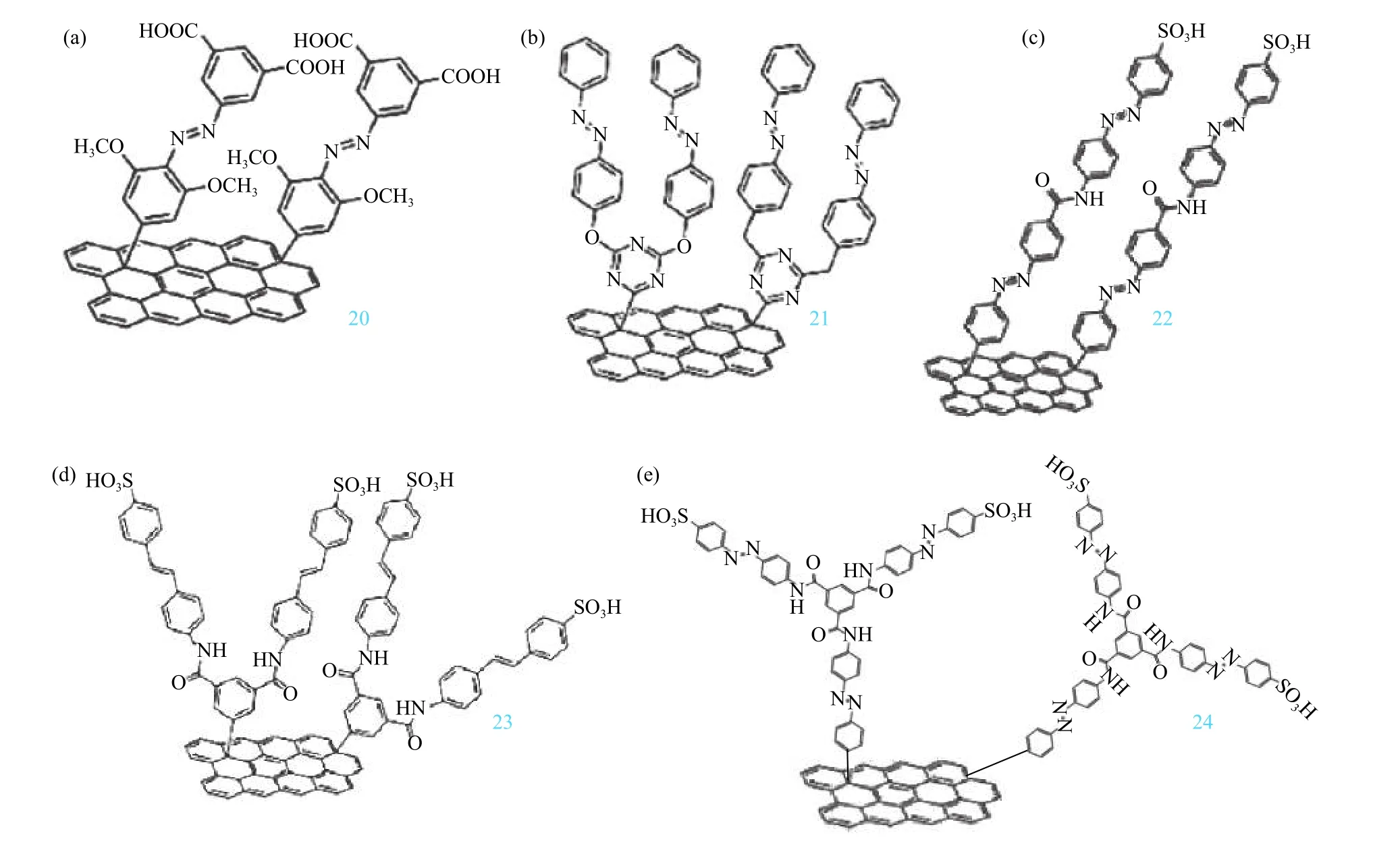
图 14 石墨烯模板化偶氮苯杂化材料化学结构[42, 78-80]Fig.14 Chemical structures of RGO–ortho-Azo, RGO–para-AZO, RGO–bis-Azo and RGO–tri-Azo hybrids [42, 78-80]
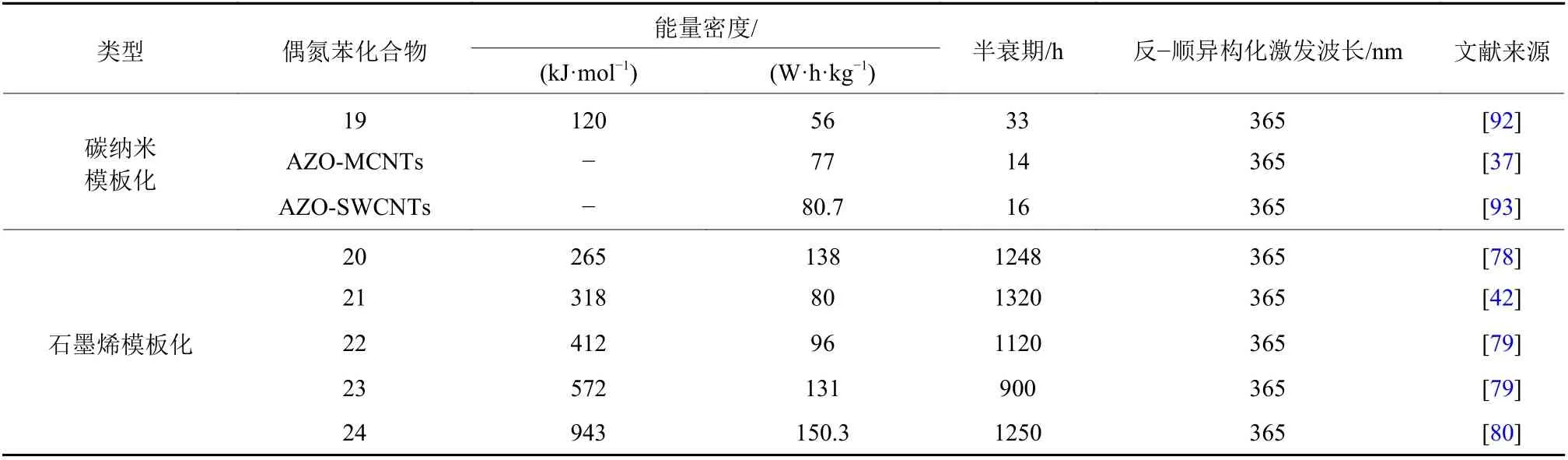
表 3 不同碳纳米模板化偶氮苯化合物的性能比较Tab.3 Comparison of properties of different carbon nano-templated azobenzene compounds
5 展 望
偶氮苯光致异构储能材料不同于其他传统的储能材料,是一个闭合循环系统,该化合物在光照条件下被激发发生光致异构化(结构改变,产生能级差)而储能,发生回复异构化而将储存的能量以热的形式释放出来,实现太阳能的储存和转化,在此储存和转化过程中不产生气体,在循环利用太阳能领域具有较大应用前景.
偶氮苯光致异构储能材料虽被证明是有前景的用于太阳能储存和转换的有效途径之一. 然而,传统的偶氮苯光致异构储能材料受到能量密度低、回复半衰期短和只能可见光储能的限制,如何实现高能长效的太阳能储存和转化和可控热释放是偶氮苯光致异构储能材料研究的重点.
(1) 毫无疑问,分子设计和微观结构优化对于实现具有高储能密度的光致异构化储能材料是至关重要的,因此,通过分子设计和结构优化开发偶氮苯光致异构化储能材料将继续是研究重点之一.
(2) 由于当前大部分文献中报道的偶氮苯光致异构储能材料不能同时满足高能量密度、长回复半衰期和能可见光储能,因此同时实现能量密度高、回复半衰期长和可见光储能仍然是一项具有挑战性的任务.
(3) 目前偶氮苯光致异构储能材料仍处于研究初期,因此,提出开发制备偶氮苯光致异构储能材料的新方法也是非常重要的.
(4) 目前对于偶氮苯光致异构储能材料用于固态膜可控热释放集中在聚合物模板化偶氮苯化合物,因此,制备碳纳米模板化偶氮苯化合物固态膜是未来研究的又一重点.
6 结语
本文通过对偶氮苯光致异构储能材料的有关论文进行综述,阐述了偶氮苯光致异构储能材料提高回复半衰期、储能密度和可用于可见光储能的方法,分析了现有研究技术的特点与不足,并在文末对偶氮苯光致异构储能材料在未来的研究方向进行了展望. 随着对偶氮苯光致异构储能材料研究的进一步深入,不仅拓宽了我们对偶氮苯光致异构储能材料的认识,还将促进其作为太阳能储能材料的多样化应用.
猜你喜欢
系统仿真技术(2022年4期)2023-01-17 13:01:44
云南化工(2021年8期)2021-12-21 06:37:38
成都大学学报(自然科学版)(2021年1期)2021-05-22 01:31:18
石油石化绿色低碳(2019年6期)2019-01-14 01:16:12
国外医药(抗生素分册)(2016年4期)2016-07-12 14:25:19
信息记录材料(2016年4期)2016-03-11 15:22:30
中国粮油学报(2016年1期)2016-02-06 02:17:09
化工进展(2015年6期)2015-11-13 00:26:50
化工进展(2015年3期)2015-11-11 09:09:44
华东理工大学学报(自然科学版)(2015年3期)2015-11-07 09:17:13
