
蛋白酶激活受体拮抗剂在抗血小板领域的研究进展
2019-10-09 02:34李杉杉朱雄
药学研究 2019年9期
李杉杉,朱雄
(中国药科大学,江苏 南京 210009)
动脉粥样硬化所形成的血栓性疾病是全世界发病率和死亡率较高的疾病之一,临床上主要表现为:急性冠状动脉综合征、脑血管事件和外周动脉疾病。其中血小板活化和聚集是导致动脉粥样硬化性疾病形成以及临床表现的主要原因[1]。目前,临床上治疗治疗血栓性疾病的标准治疗方案是阿司匹林联合P2Y12二磷酸腺苷(ADP)受体抑制剂氯吡格雷的双重口服治疗,并且此治疗方案可显著改善动脉粥样硬化疾病患者的预后[2]。然而,阿司匹林和氯吡格雷不仅抑制血小板活化途径,还可影响人体中其他的正常代谢途径[3]。并且,阿司匹林和氯吡格雷存在个体差异性和较为严重的出血事件限制了临床上的使用。凝血酶可以不断地刺激血小板的活化和血栓的形成,被认为是最强的血小板活化因子。因此,开发更有效地抑制血小板的活化而不增加出血并发症的抗血小板药物是当下血栓性疾病治疗领域存在的主要瓶颈。在临床前模型研究和小规模临床试验中发现蛋白酶激活受体(PARs)拮抗剂可抑制凝血酶介导的血小板活化,同时不增加出血风险[4]。
凝血酶(thrombin)是一种丝氨酸蛋白酶,除了促进凝血因子Ⅴ、Ⅷ、Ⅺ产生以及裂解纤维蛋白原转化为纤维蛋白外,同时也是血小板活化最强的激动剂之一[5]。凝血酶活化血小板涉及G蛋白偶联的七次跨膜受体,主要与血小板初期的聚集有关。人体血小板的活化主要是通过凝血酶介导的PAR-1和PAR-4受体进行的[6]。在人类血小板中,PAR-1是主要的凝血酶受体,PAR-1在凝血酶浓度仅有纳摩尔级时即可活化血小板,而PAR-4则需要更高浓度的凝血酶才可激活血小板[7-8]。
1 PARs及其激活机制
PARs是细胞表面的一种G蛋白偶联受体,属于G蛋白偶联受体超家族成员之一。同其他G蛋白偶联受体一样,PARs也具有单链七次跨膜的特性。近20年来,PARs共有4种蛋白酶激活受体被发现,分别是PAR-1、PAR-2、PAR-3和PAR-4。其中人类血小板表达PAR-1和PAR-4,而啮齿类动物表达PAR-3和PAR-4,而不表达PAR-1[9]。
1.1 PAR-1的生物学结构 PAR-1是最早发现的PARs家族成员,是凝血酶的主要受体,也是目前研究最多的蛋白酶活化受体。如图1所示,PAR-1位于细胞膜上,由胞外区(N-末端)、7个疏水跨膜螺旋以及胞内区(C末端)3个部分组成,共有425个氨基酸残基[10]。
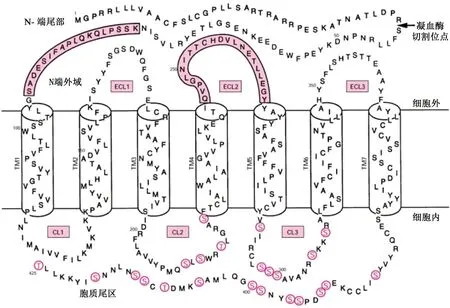
图1 人类凝血酶受体PAR-1结构[11]
1.2 凝血酶激活PAR-1机制 凝血酶含有3个不同的区域,一个活性位点和两个外结合位点。凝血酶的两端为带正电荷的外结合位点,PAR-1的N末端的酸性区能与外结合位点结合,这个酸性区含有类水蛭素的结合位点,可增强其对凝血酶的亲和力[12]。如图2所示,凝血酶的外结合位点结合血小板表面上PAR-1的水蛭素样的胞外氨基末端结构域,切割41位精氨酸和42位丝氨酸之间的受体,去除氨基末端序列并产生一个新的锁系质子化的氨基配体—SFLRRN(丝氨酸-苯丙氨酸-亮氨酸-亮氨酸-精氨酸-天冬氨酸)[13],这个新产生的N末端的锁系配体(tethered ligand)与受体发生分子内结合而产生跨膜信号从而激活PAR-1受体[14]。水蛭素样序列主要存在于PAR-1和PAR-3中,在低凝血酶浓度条件下,其对于受体的切割十分重要。而PAR-4没有促凝血酶结合的类水蛭素结合位点,因而对凝血酶的亲和力远低于PAR-1,所以PAR-4的激活需要更高的凝血酶浓度[15]。
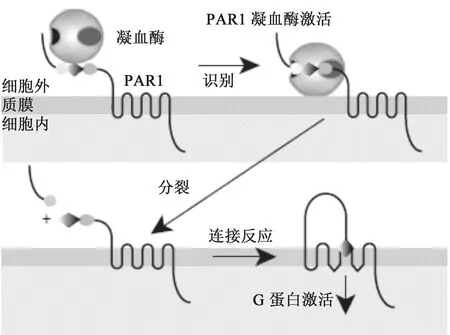
图2 凝血酶激活PAR-1机制[16]
1.3 PAR-2和PAR-3在血小板活化中的作用 PAR-2和PAR-1结构相似,含有7个跨膜螺旋结构域,N末端位于胞外,有相应受体的蛋白酶裂解位点,C末端在胞内调控受体脱敏和信号传导。然而由于PAR-2裂解位点没有PAR-1中供凝血酶结合的酸性区域,不能被凝血酶激活,而是由胰蛋白酶激活[17]。PAR-3是继PAR-1之后第二个被发现的凝血酶受体,研究发现凝血酶还可裂解PAR-3,然而这种相互作用的生理机制尚未探究清楚[18],PAR-3还可与其他PARs发生二聚化,这种行为可能有助于PAR-4的信号调节,因而PAR-3被认为是PAR-4的辅助因子,可增加凝血酶对PAR-4的亲和力[19]。
1.4 凝血酶激活PAR-4机制 PAR-4由385个氨基酸组成,包含一个信号肽和一个胞外N末端Arg/Gly/Ser蛋白酶结合位点,PAR-4的激活原理同PAR-1相似:首先PAR-4的胞外N末端与凝血酶相结合,凝血酶切割其末端并产生新的N末端即锁系配体,这个新产生的锁系配体与胞外第二环结合形成闭环,进而激活受体并引起一系列信号通路。如图3所示,但是与PAR-1有所不同,由于PAR-4没有促凝血酶结合的类水蛭素结合位点,需要较高浓度的凝血酶PAR-4才可激活。当PAR-3和PAR-4同时表达时,在较低浓度的凝血酶条件下PAR-3可作为辅助因子提高PAR-4的敏感度,有助于PAR-4的裂解和活化[20]。
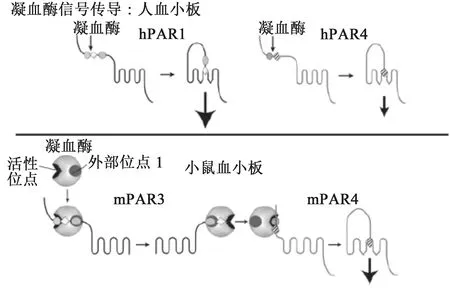
图3 凝血酶激活PAR-4以及辅助因子PAR-3参与的调节机制[19]
2 PAR-1和PAR-4作为抗血小板聚集治疗靶点的优势
2.1 现有临床药物的局限性 目前临床上心脑血管疾病标准的治疗方案是阿司匹林联合氯吡格雷的双重口服的抗血小板治疗。阿司匹林是不可逆的COX-1抑制剂,通过减少TXA2的生成从而抑制血小板过度聚集,达到防止血栓生成的目的[21]。氯吡格雷通过与P2Y12不可逆结合,抑制P2Y12受体活性,从而抑制血小板聚集[22]。两种药物联合使用能够有效地抑制血小板聚集,防止血栓的生成,降低心脑血管的发病率。但是该治疗方案可以引起严重的出血事件,并且出血风险随剂量的依赖性而增加;其次除了出血风险之外,相当多的患者在接受双重抗血小板治疗之后更容易产生反复的血栓形成事件[23]。因此对于具有强效和低出血风险的抗血小板药物仍存在大量的临床需求。
2.2 PARs拮抗剂的优势 凝血酶介导的人类血小板活化的过程中,PAR-1和PAR-4发挥着明显不同的作用。PAR-1对凝血酶的亲和力高,激活所需要的凝血酶浓度较低;相反PAR-4是一种低亲和力的凝血酶受体,需要较高浓度的凝血酶来激活。PAR-1和PAR-4在人体内的动力学过程不同,并且参与了血小板活化的不同阶段。凝血酶激活PAR-1产生快速而短暂的钙离子信号[24-25],进而引发受体的再分配和信号传导的终止,而PAR-4的活化速度比PAR-1慢20~70倍,并且所引发的钙信号持续时间较长[26]。有理论研究认为,在凝血酶浓度较低的初始情况下,PAR-1的激活是参与止血过程的主要因素;而在高凝血酶浓度的条件下血小板活化的后期阶段以闭塞性血栓的形成为主,这个过程主要是由PAR-4介导完成的[27]。PARs拮抗剂并不影响凝血酶切割纤维蛋白酶的功能,相比于其他的凝血酶抑制剂,PARs拮抗剂不容易引起出血并发症,且具有更为宽阔的治疗窗[28]。因此PAR-1和PAR-4作为靶点开发新的抗血小板聚集药物具有较大的前景,如表1所示,PAR-1和PAR-4拮抗剂的临床研究进展。
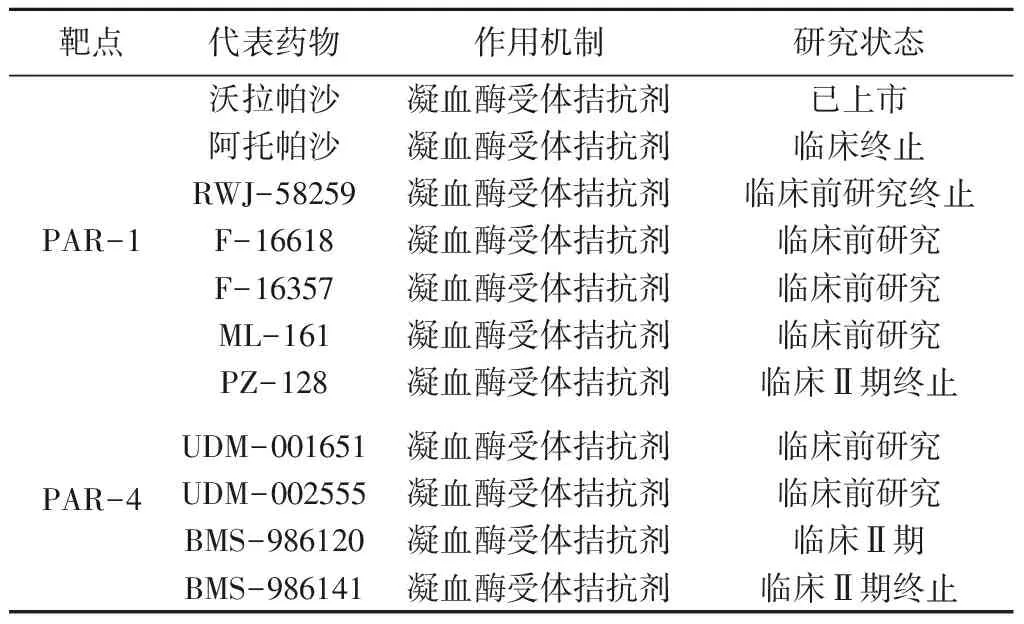
表1 PAR-1和PAR-4拮抗剂临床研究进展
3 PAR-1拮抗剂的研究进展
PAR-1拮抗剂:如图4所示,目前报道的PAR-1小分子拮抗剂主要包括Vorapaxar、Atopaxar、F-16618、F-16357、ML-161、RWJ-52859、PZ-128等。
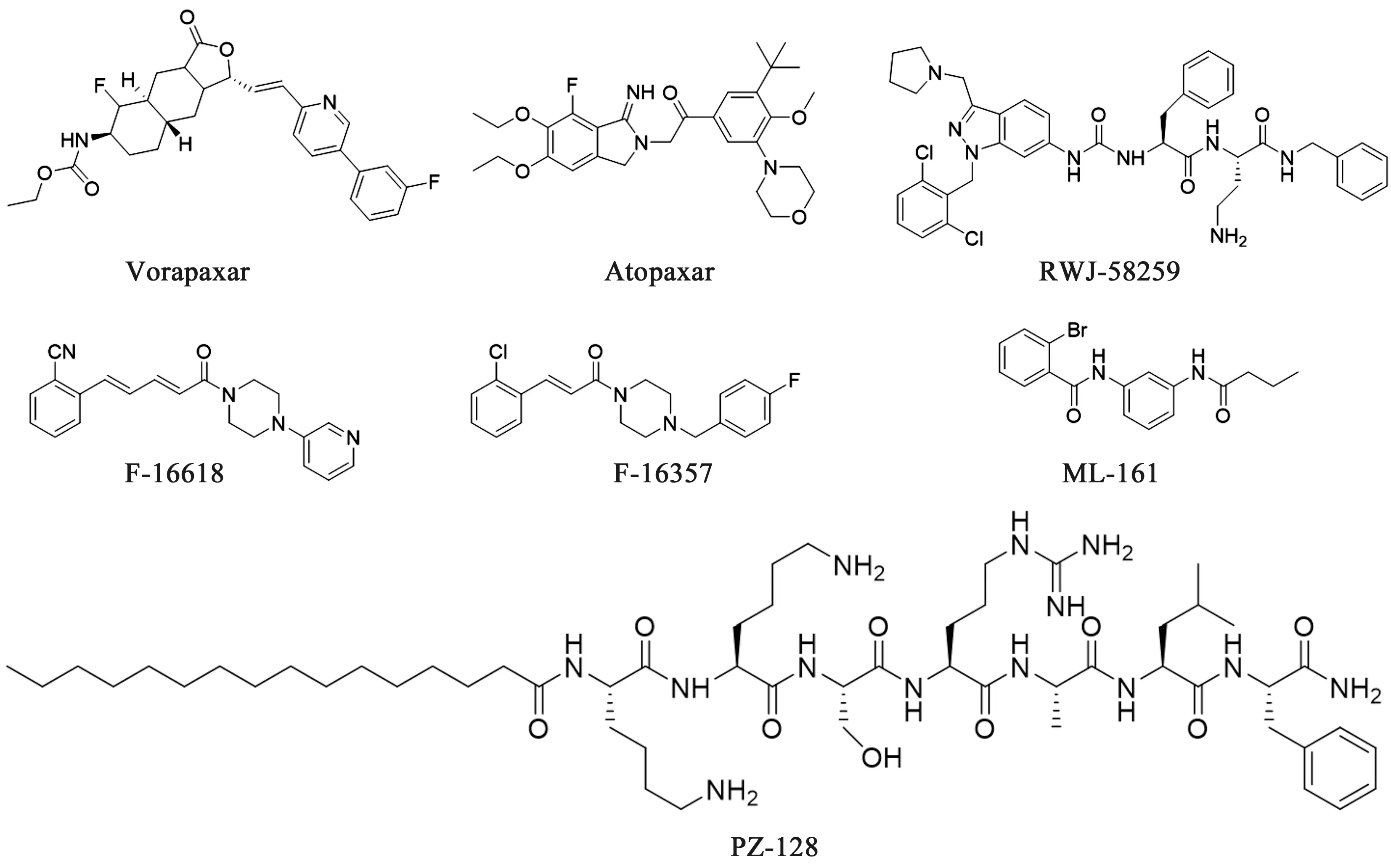
图4 PAR-1拮抗剂
3.1 Vorapaxar Vorapaxar由默沙东研发,2014年5月经FDA批准上市,是首个通过抑制凝血酶受体的活性来治疗血栓的药物。研究显示,Vorapaxar对PAR-1具有良好的拮抗活性,能显著降低心肌梗死的发病率。然而当患者患有中风史或者短暂性脑缺血发作史时,Vorapaxar会增加颅内出血的风险[29]。而且没有合适的可逆转Vorapaxar的抗血小板的药物,2016年8月,默沙东宣布停止在美国继续推广Vorapaxar的临床使用。
3.2 Atopaxar Atopaxar又名E5555,由日本卫材株式会社研发的可逆性口服PAR-1拮抗剂。临床前研究表明:Atopaxar可有效抑制凝血酶诱导的血小板聚集反应(IC50=64 nmol·L-1),表现出强有力的血小板抑制作用,同时不会增加出血时间[30]。然而在Ⅱ期临床的高剂量组试验中,发现了Atopaxar可引起肝功能异常和QT间期延长的现象,并且缺乏明显的量效关系,故已终止了Atopaxar的开发[31]。
3.3 RWJ-58259 RWJ-58259由强生公司研发的选择性PAR-1拮抗剂。在体外活性测试中,发现RWJ-58259可有效抑制人体凝血酶介导的血小板聚集(IC50=0.37 μmol·L-1)[32]。然而药动学性质较差使其止步于临床前研究。
3.4 F-16618、F-16357 F-16618、F-16357是法国Pierre Fabre 研究中心开发的PAR-1 拮抗剂。体外研究表明F-16618、F-16357可有效抑制凝血酶受体活性肽诱导的血小板聚集,并且在大鼠的体外动静脉模型中均表现出一定的抗血栓活性,对PAR-1有一定选择性[33]。F-16618、F-16357药动学性能良好,目前尚未有更多的临床信息披露。
3.5 ML-161 ML-161是由Dockendorff等[34]通过高通量筛选发现的新型PAR-1拮抗剂。临床前研究表明ML-161能够显著抑制由凝血酶受体活性肽诱导的人血小板表面P-selectin 蛋白的表达,且仅抑制了血小板的分泌反应,而不改变血小板的正常形态。这种新的作用机制可能降低临床上出血等不良事件的发生,目前尚未有更多临床信息披露。
3.6 PZ-128 PZ-128由Tufts Medical Center开发的高效、可逆转的多肽类PAR-1拮抗剂,I期临床显示PZ-128对由凝血酶受体活性肽(8 μmol·L-1)诱导的血小板聚集反应具有明显效果,并且呈剂量相关性[35]。2018年4月24日披露信息显示其暂停了一项名为“PZ-128在接受非急诊经皮冠状动脉介入治疗(TRIP-PCI)的患者中的安全性”的Ⅱ期临床实验,暂停原因尚不明确。
4 PAR-4拮抗剂研究进展
近二十年来,围绕PAR-4特有的生理机制,研究人员通过生物电子等排、高通量筛选等技术设计合成了3种不同结构类型的小分子的PAR-4拮抗剂,主要包括吲唑、吲哚、咪唑并噻二唑类结构。
4.1 吲唑类 2001年,我国台湾的研究工作者Lee等[36]发现YC-1对由凝血酶、花生四烯酸、胶原蛋白以及血小板活化因子引起的血小板聚集具有显著的抑制作用[37]。如图5所示,在YC-1的基础上设计合成了一系列吲唑苯甲酸类化合物,并对其进行了活性筛选。如改造的过程中发现YD-3对由凝血酶介导的血小板聚集有很强的抑制作用(IC50=29.3 μmol·L-1)[38],而对花生四烯酸、胶原蛋白、血小板活化因子等没有抑制作用。YD-3可以选择性地抑制凝血酶诱导的血小板聚集和磷酸肌醇的分解而不影响凝血酶正常的蛋白水解活性,研究人员在深入的研究中发现YD-3并非是PAR-1受体拮抗剂,而是一种非肽类的PAR-4受体拮抗剂。然而由于YD-3的高亲脂性限制了其在体外实验中的应用。由此研究人员以YD-3作为先导化合物对其进行结构改造和构效关系的研究。
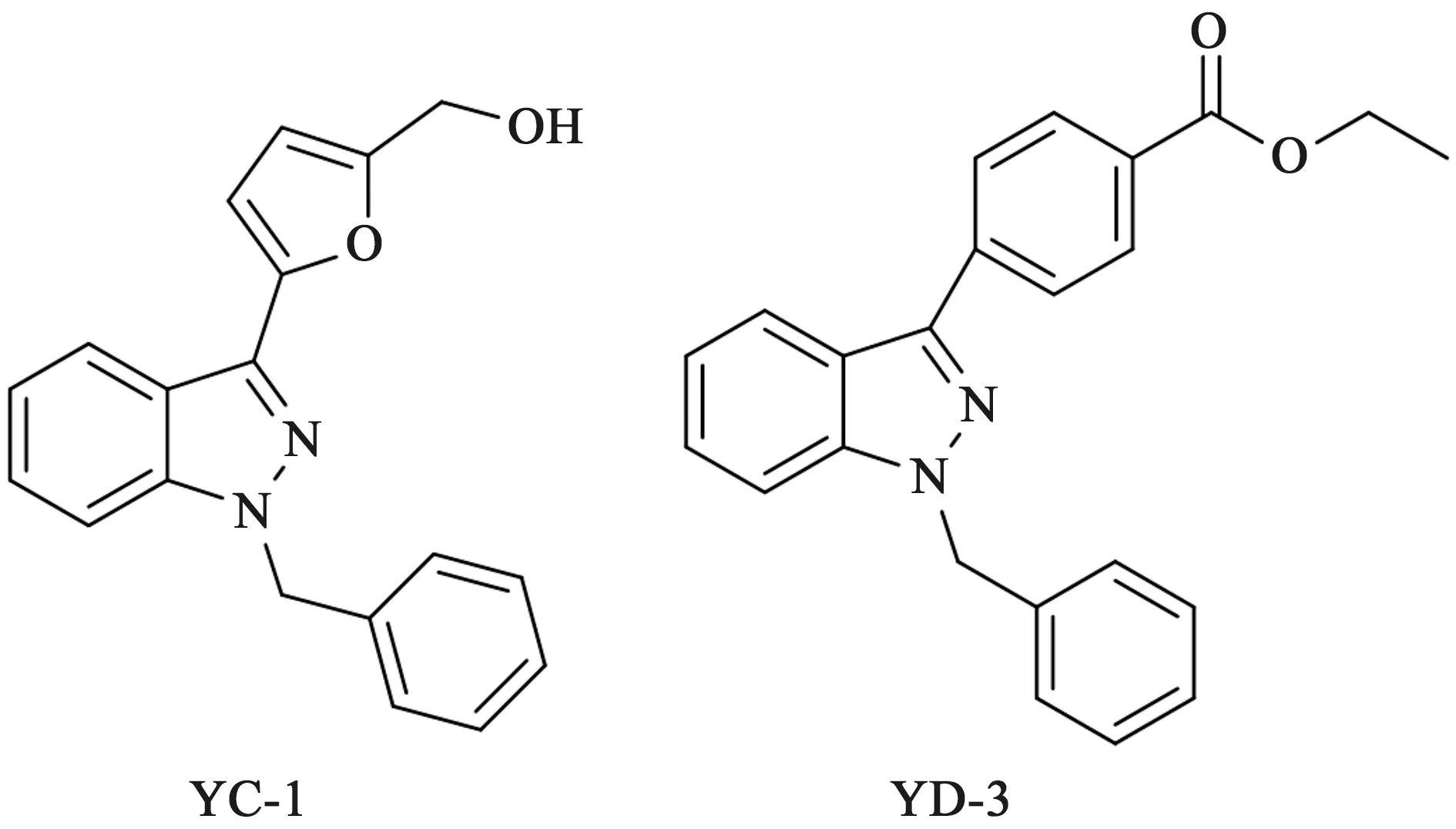
图5 YC-1和YD-3的化学结构
2008年,研究者在对YD-3进行构效关系的探索中发现[39],C3位苯环上的乙酯基和N1位上的苄基对于抗PAR-4活性是重要的药效基团,二者缺少其中的任一部分均可导致活性丧失;C3位上的乙酯基被其他酯、酰胺或者羧酸基团取代时活性降低;N1位上苄基引入卤素或者甲氧基可增加对PAR-4受体的选择性。
4.2 吲哚类 2013年,Wen等[29,40]在YD-3的基础上将吲唑环改为吲哚环,设计合成了比YD-3活性更好的化合物SEY-3(IC50=66 nmol·L-1),并在Vanderbit大学的化合物库中进一步筛选出与SEY-3结构类似的化合物ML-354(IC50=140 nmol·L-1)。由于毒理学和药代动力学等方面的因素,研究者继续以ML-354为先导化合物进行结构修饰和构效关系的研究,化合物结构如图6所示。
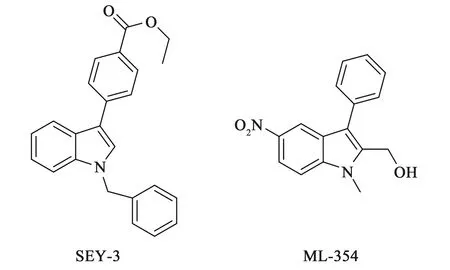
图6 化合物SEY-3和ML-354化学结构
同时,Temple等[41-42]在ML-354的基础上也引入了咪唑并噻二唑的结构,得到了更优的活性和体内药代动力学参数,并且对γ-凝血酶(γ-thrombin)有很好的抑制作用。研究发现VU0661259(IC50=179 nmol·L-1)的药理活性比由ML-354修饰而来的化合物1活性更好,在结构修饰以及寻找最小药效团的过程中,研究者将VU0661259 的C2位引入咪唑并噻二的结构来替代羟甲基的结构,得到了对PAR-4 活性肽(PAR-4 Activating Peptide,PAR-4 AP) IC50=20 nmol·L-1的化合物2,化合物结构如图7所示。这些工作为后期的具有成药性的化合物提供了重要的借鉴意义。
4.3 咪唑并噻二唑类 2013年,施贵宝公司(Bristol-Myers Squibb,BMS)也对PAR-4拮抗剂展开了大量的研究工作并申请的大量的专利[43-45]。在对大量咪唑并噻二唑化合物的合成和筛选过程中,发现了UDM-001651、UDM-002555、
BMS-986141、BMS-986120(见图8)等化合物对PAR-4引起的血小板聚集具有优良的抑制作用。
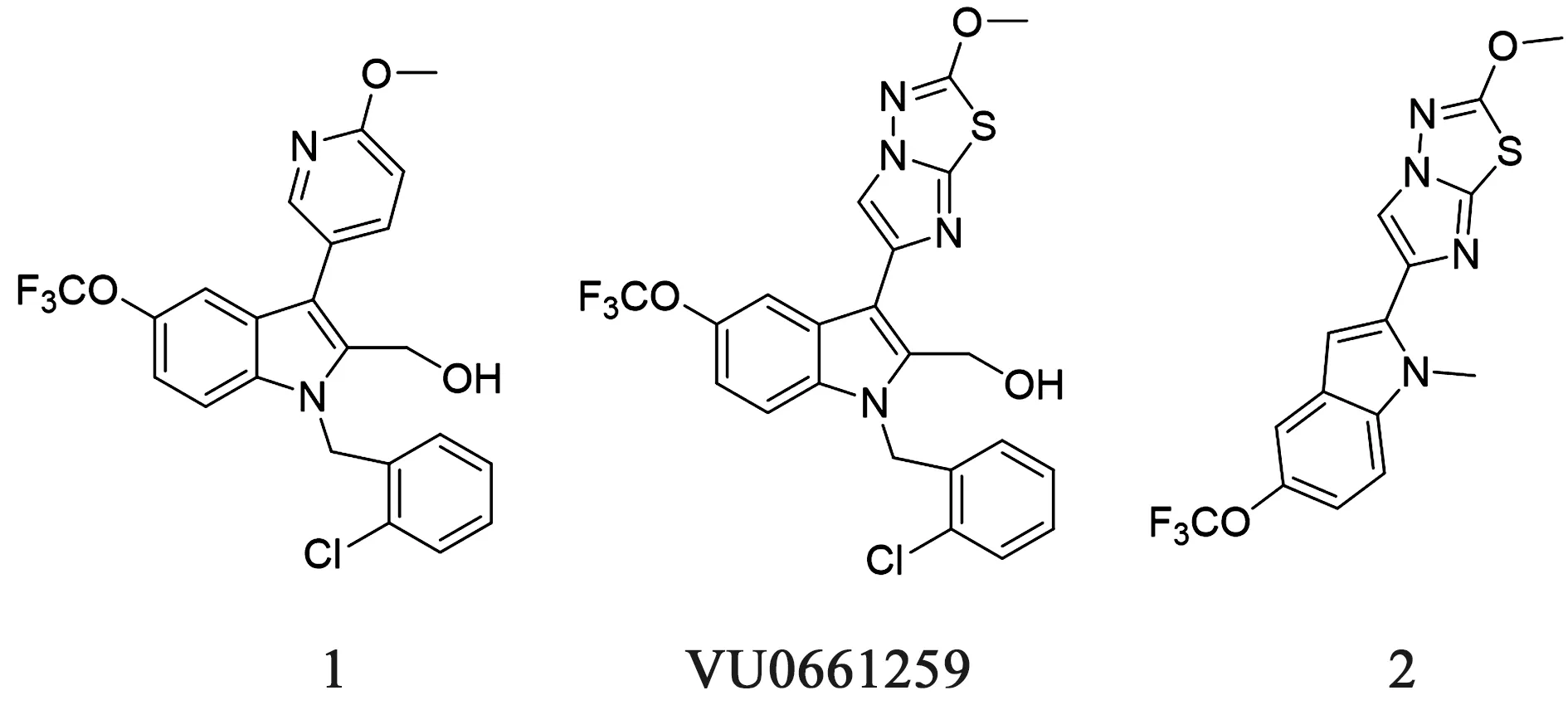
图7 化合物1~2、VU0661259化学结构
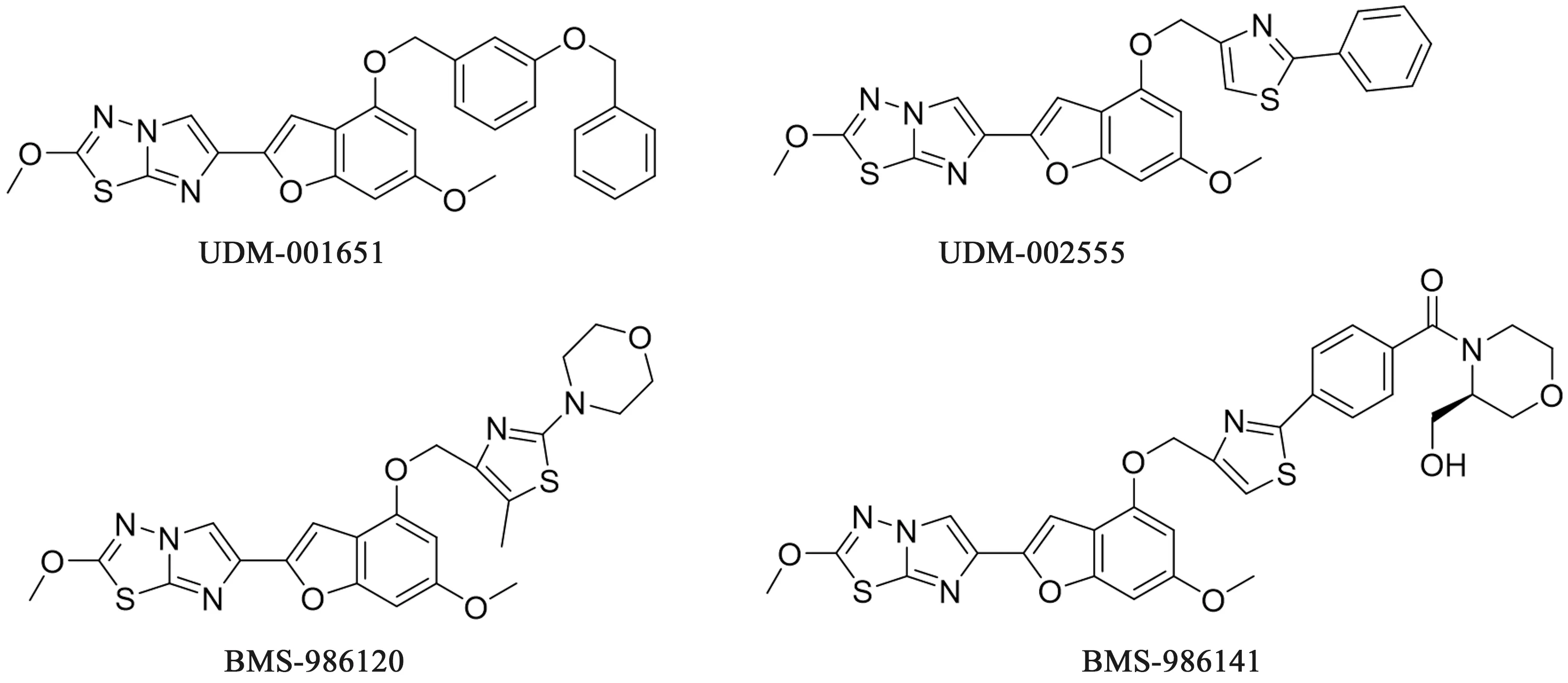
图8 BMS开发的PAR-4拮抗剂化学结构
UDM-001651和UDM-002555均已完成临床前研究,其中UDM-001651和UDM-00255的体外研究中对于拮抗人体PAR-4 AP IC50值分别为2.42和1 nmol·L-1,目前已作为一种工具药物检测PAR-4激动肽的活性。
BMS-986120是一种口服强效、可逆、选择性好的小分子PAR-4受体拮抗剂,BMS-986120可选择性地抑制PAR-4受体,而对一系列凝血蛋白酶和多种受体、酶以及离子通道均无抑制活性。在体外实验中,BMS-986120对γ-凝血酶的IC50=7.3 nmol·L-1,而在人类全血中的PAR-4 AP IC50=9.5 nmol·L-1。BMS-986120已完成Ⅰ期临床实验,临床结果表明BMS-986120具有很强的剂量依赖性的抗血栓活性,且药代动力学数据可接受,相比于标准抗血小板药物氯吡格雷BMS-986120表现出较低的出血风险和更宽的治疗窗口。目前BMS-986120已进入Ⅱ期临床。
BMS-986141也是口服小分子PAR-4受体抑制剂,用于预防急性缺血性脑卒中或接受阿司匹林治疗后的短暂性脑缺血以及复发性的脑梗死[46]。其体外对血小板聚集反应的IC50达0.33 μmol·L-1。2017年在Ⅱ期临床中因为存在较高比例的不良反应已终止开发。
5 总结与展望
近年来,抗血小板药物的开发已成为当下心脑血管疾病领域的研究热点。虽然出现了各种不同机制的抗血小板药物,但临床上不良反应的发生率仍然很高。PAR-1和PAR-4已成为抗血小板药物研发的新靶点,目前PAR-1受体拮抗剂Vorapaxar虽已上市,但出血事故的发生比例较高,限制了其在临床上的应用。近期研究发现药物靶向拮抗PAR-4受体时,发生出血的不良反应较低,针对PAR-4的小分子拮抗剂有可能成为安全有效的抗血小板药物,具有广阔的市场前景。但PAR-4受体拮抗剂存在药代动力学参数较差,成药性不好等问题。目前仅施贵宝公司研发的药物BMS-986120进入临床Ⅱ期。相信随着对PAR-1和PAR-4靶点的不断深入以及相关拮抗剂结构的优化,抗血小板治疗领域将会取得巨大的突破。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
云南化工(2020年11期)2021-01-14
作文成功之路·小学版(2020年6期)2020-07-27
天然产物研究与开发(2018年10期)2018-11-06
中华老年多器官疾病杂志(2016年7期)2016-04-28
中国社区医师(2015年14期)2015-12-24
医学研究杂志(2015年12期)2015-06-10
中华皮肤科杂志(2014年4期)2014-12-19
分析化学(2014年7期)2014-12-13
食品工业科技(2014年5期)2014-03-11
