
RP-HPLC法测定载替加环素脂质微泡的包封率
2019-10-09 02:33任雅君王海翔王岁楼徐艳艳田吉来
药学研究 2019年9期
任雅君,王海翔,王岁楼*,徐艳艳,田吉来
(1.中国药科大学工学院,江苏 南京 210009;2.丽水市中心医院临床药学室,浙江 丽水 323000; 3.南京中医药大学医学与生命科学学院,江苏 南京 210023)
替加环素(Tigecycline)为甘氨酰环素类抗菌药,半合成四环素米诺环素的衍生物。为第一个批准上市的甘氨环素类抗菌药物。该药克服了四环素的两种主要耐药机制:药物特异性外排泵和核糖体保护。替加环素对许多革兰阳性和阴性细菌有效,包括耐甲氧西林金黄色葡萄球菌、万古霉素耐药的肠球菌以及产超广谱β-内酰胺酶大肠埃希菌和肺炎克雷伯菌。其通过与核糖体30S亚单位结合、阻止氨酰化tRNA分子进入核糖体A位而抑制细菌蛋白质合成。因其不受β内酰胺酶、靶位修饰和酶靶位改变等耐药机制的影响,具有广谱抗菌活性[1]。但由于替加环素组织分布广、蛋白结合率高、脑脊液浓度低、临床常规治疗多重耐药鲍曼不动杆菌(MDRAB)颅内感染疗效差的问题,其临床疗效受到限制[2]。
超声联合微泡技术(ultrasound combining microbubbles,USCM)被证实可以有效、安全和可逆地打开血脑屏障,是一种理想的实现药物靶向性、无创地进入颅内组织的技术方法[3-4]。其原理是将超声微泡造影剂(ultrasound contrast agents,UCA)作为载体,将药物包载于微泡中,后通过超声辐照定点靶向释放药物达到治疗的目的[5-6]。具体来说,即静脉注入载药微泡后,用超声辐照特定部位,含气体的超声造影剂在超声波的作用下会不断地产生非对称性收缩和膨胀,当声能达到一定强度时,微泡就会破裂,这样药物就被释放到超声所辐照的局部组织中,从而发挥定位靶向治疗作用。目前,测定替加环素含量多用反相液相色谱法:Li等[7-8]等均采用C18柱、磷酸缓冲液-乙腈(72∶25)作为流动相测定替加环素含量,出峰时间8 min左右,均获得良好的线性及回收率;吉同琴等[9-10]采用C18柱、乙酸铵溶液(含EDTA和四氢呋喃以及三乙胺调pH 7.9)为流动相,出峰时间15 min左右,线性与回收率良好。
本试验制备了不同药脂比的载替加环素脂质微泡,通过查阅大量文献,优化建立新型高效液相色谱(HPLC)法测定其包封率,以筛选出最佳药脂比,同时证实了微泡与药物联用的可行性,为后期超声辐照靶向释放药物试验奠定了基础。
1 仪器与试药
1.1 仪器 LCsolution 15C型高效液相色谱仪(日本岛津公司);AX224型分析天平(日本岛津公司);FD-1C-50型真空冷冻干燥机(北京博医康实验仪器有限公司);YJ-100型银汞胶囊调合器(杭州新亚光电仪器有限公司);恒温磁力搅拌器(美国Scilogex公司);TGL-16G型冷冻超速离心机(上海安亭科学仪器厂);pH测定仪(上海雷磁仪器厂)。
1.2 试药 二硬脂酰磷脂酰胆碱(DSPC,苏州东南药业股份有限公司);1,2-棕榈酰磷脂酰甘油钠(DPPG-Na,苏州东南药业股份有限公司);生物素化磷脂(DSPE-PEG2000-Biotin,Aladdin公司);替加环素标准品(99%,批号:291641,北京百灵威科技有限公司);SF6(99.999%,南京上元工业气体厂);pH=7.4磷酸盐缓冲液(PBS,自制);甘油、叔丁醇、草酸、磷酸二氢钠、EDTA-2Na、三乙胺均为分析纯,乙腈、甲醇均为色谱纯。
2 方法与结果
2.1 载药微泡的制备
2.1.1 空白脂质微泡的制备 称取一定量DSPC、DPPG-Na、DSPE-PEG2000-Biotin溶于20 mL叔丁醇,再加入适量PEG4000和少量棕榈酸,60 ℃水浴溶解60 min;冷冻干燥30 h;取冻干粉一瓶加入1 mL PBS/甘油水合液45 ℃水化30 min,4 ℃静置20 min,再水化30 min,室温放置。磷脂浓度为4 mg·mL-1。充入SF6气体,银汞胶囊调和器振荡90 s。空白脂质体制备完成,静置分层,上层乳白色泡沫状,下层清液。4 ℃保存。
2.1.2 载替加环素脂质微泡的制备 成膜材料及辅料同空白微泡,再分别精密称取4、2和1 mg的替加环素,用适量叔丁醇溶解后,缓慢加入磷脂溶液中,60 ℃水浴搅拌1 h。其余步骤同“2.1.1”项下。即制得药脂比分别为10∶1的T1组、20∶1的T2组和40∶1的T3组三组载药微泡。载药微泡静置分层,上层呈黄绿色泡沫状,下层清液。4 ℃保存。
2.2 色谱条件
2.2.1 色谱柱 Welch XB-C18柱(4.6 mm× 150 mm,3 μm);流速:1 mL·min-1;柱温:25 ℃;进样体积:20 μL。
2.2.2 流动相 磷酸盐缓冲液[(0.015 mol·L-1的磷酸二氢钠溶液和0.015 mol·L-1的草酸以及0.002 mol·L-1的EDTA-2Na(三乙胺调pH 7.0)]-乙腈溶液(75∶25)。使用前过0.45 μm有机滤膜。
2.2.3 检测波长 在190~400 nm范围内进行光谱扫描,得到替加环素的两个较大吸收峰出现在248.1 nm和357.7 nm处,因在248 nm处的吸收更灵敏,响应值更大,故选择248 nm作为最大吸收波长。
2.3 标准品溶液配置 精密称取替加环素标准品5 mg超声溶解于25 mL流动相中,制成0.2 mg·mL-1标准品储备液。4 ℃保存。使用前过0.45 μm有机滤膜。取20 μL进样测定,观察保留时间、峰形与拖尾因子,结果如图1。
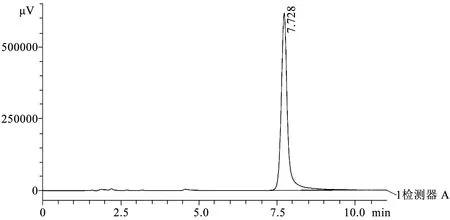
图1 替加环素标准溶液0.2 mg·mL-1色谱图
3 方法学验证
3.1 标准曲线绘制 取储备液按照一定比例稀释,配制成5、10、20、40、80、100、200 μg·mL-1一系列浓度标准品溶液,HPLC进样测定,外标法定量。 以峰面积(UV)为纵坐标,浓度(μg·mL-1)为横坐标做线性回归。由结果可知,替加环素标准品200 μg·mL-1的色谱峰峰形良好,保留时间为7.728,理论塔板数为8 663.2,拖尾因子为1.2,无其他杂质峰。
标准曲线的线性回归方程为Y=44 468X-121 566,R2=0.999 5。说明替加环素在5~200 μg·mL-1的浓度范围内线性良好。
3.2 重复性测试 配6份40 μg·mL-1替加环素标准品溶液,分别进样测定。计算RSD值,结果如表1。由结果可知,6份相同浓度的标准品进样结果的RSD=2.2%,说明该法重复度较好。
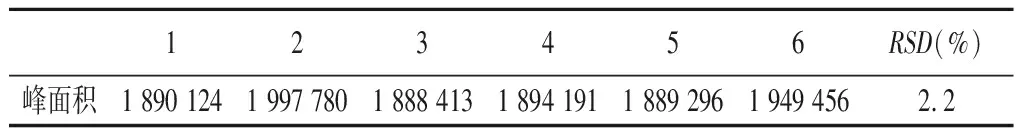
表1 RP-HPLC法测定替加环素含量的重复度试验
3.3 精密度测定 取40 μg·mL-1标准品溶液连续测定6次,计算RSD(%),结果如表2。由结果可知,连续6次进样结果的RSD为2.1%,说明该方法精密度较好。
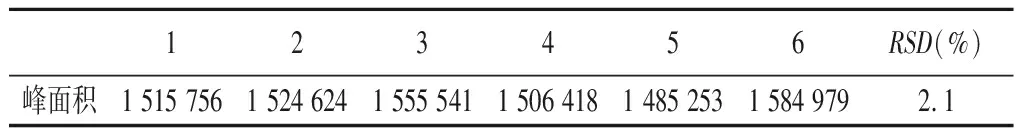
表2 RP-HPLC法测定替加环素含量的精密度试验
3.4 加标回收率测定 取9份0.2 mL空白脂质微泡溶液,分别加入0.08、0.1、0.12 mg替加环素标准品。流动相定容至1 mL,配置成80、100、120 μg·mL-13种浓度加标待测液,每种3个平行,HPLC测定峰面积,计算浓度、回收率及RSD(%),结果如表3。
由试验结果可以看出,高、中、低3个浓度水平组的加标回收率分别为104.3%(RSD=0.8%)、100.3%(RSD=1.5%)、99.6(RSD=0.4%)。说明在本试验条件下替加环素的回收率良好,辅料(空白脂质微泡)对主药(替加环素)的测定无影响。该方法测定替加环素的含量可行。
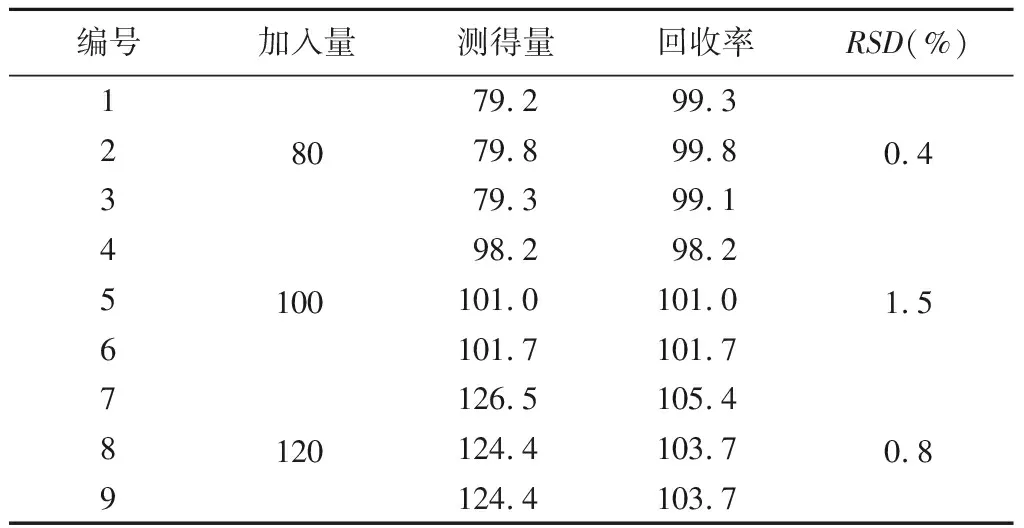
表3 加标回收率试验结果
3.5 包封率测定 采用高速离心沉淀法:混匀包裹的新鲜载药微泡溶液样品 1 mL,12 000 rpm离心 10 min后取下清液,过0.45 μm滤膜后进样,测定游离的替加环素含量。每种样品测定3次。
包封率(%)=(投入的替加环素总量-游离的量)/投入的替加环素总量×100%
载药量(%)=(投入的替加环素总量-游离的量)/磷脂用量×100%
由表4及图2所示,T1组、T2组和T3组的包封率分别为54.3%±4.9%、86.8%±0.6%和71.3%±1.6%;载药量分别为5.4%±0.5%、4.3%±0和1.8%±0。三者的包封率都较为理想,T2组即药脂比为20∶1的载药微泡的包封率最大,T1组即药脂比为10∶1的载药微泡的包封率最小。因此,最终选择药脂比为20∶1作为载药微泡的制备条件。

表4 3组载药微泡包封率与载药量测定结果
4 讨论
4.1 流动相的选择 替加环素是四环素的结构类似物,拥有该类化合物相似的化学特性。同时为含氮碱性药物,极性较大,可以与金属离子形成金属螯合物,并吸附在反相色谱柱中的硅烷醇基上而难以洗脱,从而导致色谱峰拖尾[11]。此外,硅胶中的微量金属可以导致硅羟基的酸性增强,也会导致主峰拖尾[12]。
为了消除金属离子的影响,提高流动相的洗脱性,本试验考察了磷酸、草酸、三乙胺以及EDTA-2Na的加入对色谱峰值的影响,包括拖尾因子、保留时间以及理论塔板数。试验证明,草酸的效果较磷酸更好,三乙胺和少量EDTA-2Na的加入能够改善峰形、提高峰形的对称性、降低拖尾因子。因此最终选择了磷酸缓冲液(加入适量草酸与EDTA-2Na,并用三乙胺调节pH至7.0)-乙腈(75∶25)为流动相,此时的替加环素色谱峰峰形理想,定量准确。
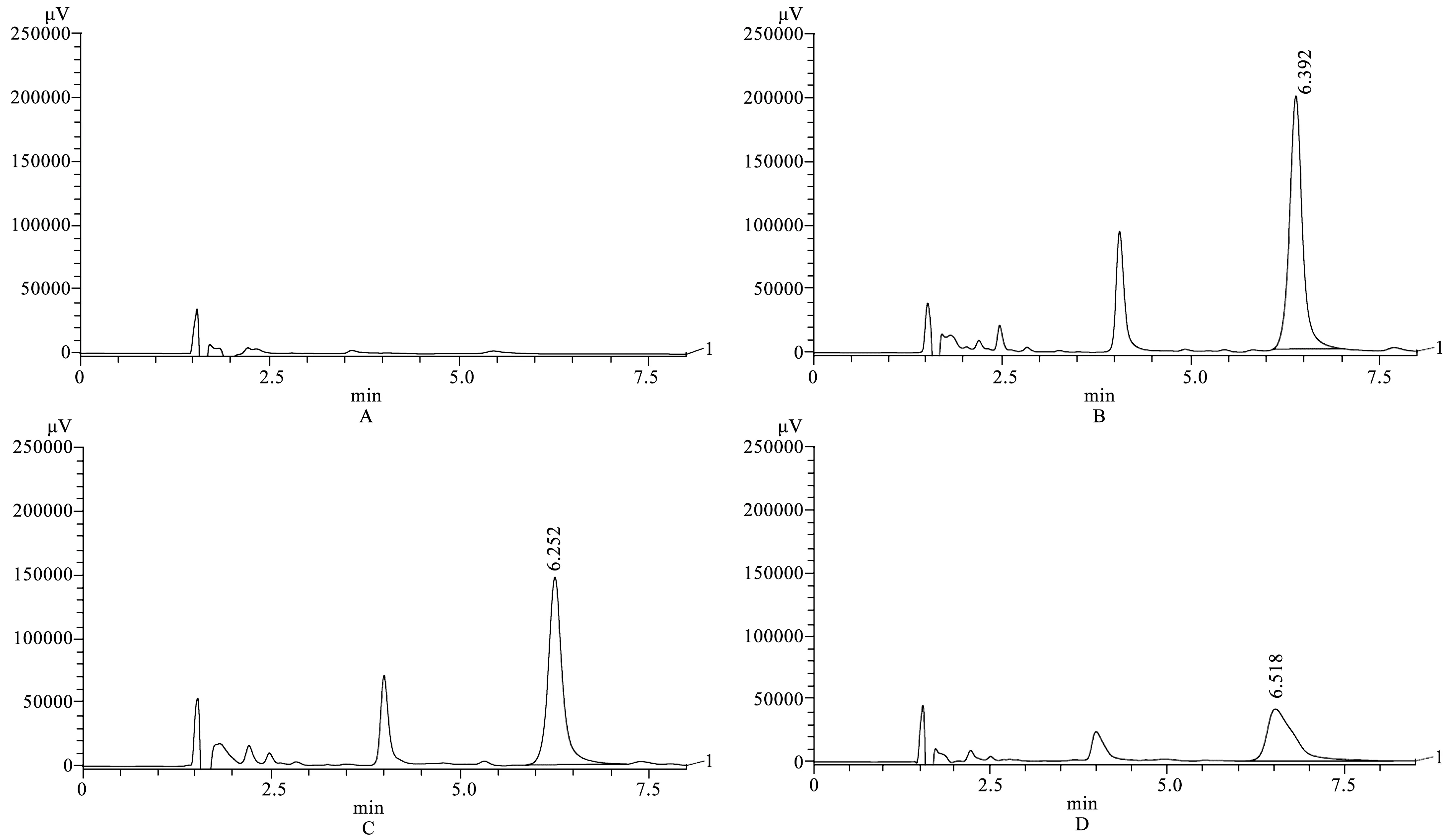
A.空白样品色谱图;B.T1组色谱图;C.T2组色谱图;D.T3组色谱图图2 不同样品色谱图
4.2 溶剂的选择 由于替加环素在水中溶解度较好,试验考察了以水作溶剂和以流动相作溶剂的替加环素色谱峰,发现流动相作溶剂的色谱峰拖尾因子更小,理论塔板数更大,峰形更理想。原因可能是流动相具有更接近中性的pH,而替加环素在中性溶液中稳定性更好。因此采用现配现用的流动相作为溶剂和稀释剂。
4.3 包封率测定方法的选择 测定载药微泡包封率的方法有很多种,如超速离心法、凝胶层析法、超滤膜过滤法、微型柱离心法和透析法等[13]。凝胶层析法、超滤膜过滤法、微型柱离心法均较烦琐、耗时长、原材料较贵,对脂膜或药物有一定吸附,导致检测误差增大,而且大小为微米级的微泡经分离柱洗脱时很难保证顺利通过以及不被破坏,而透析法会因多次换液透析后,测算相对困难,因此,本试验选择了超速离心法作为载替加环素微泡包封率的检测方法,此方法快速准确、操作简便。通过离心将未包封进微泡的药物滤出,测定其中药物含量,再由投药总量计算包封在微泡内的药物含量。此为间接计算包封率的方法,结果显示本试验制备工艺下脂质微泡有较为理性的包封率。
猜你喜欢
化工学报(2021年8期)2021-08-31
当代水产(2021年3期)2021-07-20
昆明医科大学学报(2021年2期)2021-03-29
中国医学影像技术(2018年8期)2018-08-21
中国临床医学(2016年3期)2016-07-25
天然产物研究与开发(2016年6期)2016-06-05
中国病理生理杂志(2015年8期)2015-12-21
现代检验医学杂志(2015年1期)2015-02-06
中国药业(2014年17期)2014-05-26
天然产物研究与开发(2014年6期)2014-04-27
