
从信号交联角度探讨骨髓脂肪化在原发性骨质疏松中的作用机制
2019-09-27 05:48甘东浩陈德强谭国庆徐展望
中国骨质疏松杂志 2019年8期
甘东浩 陈德强 谭国庆* 徐展望*
1. 山东中医药大学,山东 济南 250355 2.山东中医药大学附属医院骨科,山东 济南 250014
骨质疏松症(osteoporosis,OP)是以骨量减少、骨组织微细结构改变为特征,伴随有骨的脆性增加以及易于发生骨折的一种全身性代谢性骨骼疾病,常发生于老年人和绝经后妇女中。近年来,在原发性骨质疏松(老龄或绝经后)的哺乳动物和人类骨髓中脂肪增多伴骨量减少的现象引起了研究者的广泛关注,但具体机制如何,尚未完全阐明。本文将从FoxO-β-catenin-PPARγ信号轴角度对骨髓脂肪化在原发性骨质疏松中的作用机制进行综述,为该疾病的诊治提供新的着眼点。
1 原发性骨质疏松骨量减少与骨髓脂肪增多的关系
骨髓间充质干细胞(bone marrow mesenchymal stem cells,BMSCs)具有多向分化能力[1]。作为骨髓中成骨细胞和脂肪细胞的共同来源,有研究指出[2]在某些生理和病理状态下,由于骨髓内脂肪细胞数目不断增多,骨髓腔内微环境发生改变,导致具有向成骨和成脂分化潜能的BMSCs向脂肪细胞分化增多、成骨分化减少,从而打破了骨吸收和骨形成之间的平衡并形成恶性循环,最终导致或加剧了骨质疏松。体外细胞培养研究也证实来自骨质疏松大鼠的BMSCs(O-MSCs)比对照组的BMSCs(C-MSCs)有着更强的成脂分化能力,相反成骨分化能力减弱。现代研究[3]表明,在各种原发性的骨质疏松患者中,其骨髓腔内的MSCs成脂增多伴成骨能力下降,且脂肪细胞含量与骨质疏松程度呈反比,进一步说明了骨髓中脂肪细胞与成骨细胞之间存在着一种“此消彼长”的关系[4],并且已经被认为是原发性骨质疏松症的一种发病机制。那么,这种BMSCs定向分化所形成新的脂肪细胞和脂肪难道仅仅起着填充髓腔和骨小梁空隙的作用,还是会影响成骨,造成骨质疏松?一些研究[5]已经进一步指出,这些脂肪和骨有着一种密切的生理病理相关性,原发性骨质疏松骨量的丢失是脂肪和骨相关联的结果,是一种骨肥胖。骨质疏松是一种脂毒性疾病,过多沉积的脂类可以引起成骨细胞和骨细胞的凋亡及功能障碍[6]。但对这种脂毒性的具体机制研究较少,有待于进一步深入研究。
2 骨髓脂肪化对局部微环境的影响
脂毒性是指脂质在非脂肪组织过载所引起的细胞功能障碍和所诱导的细胞凋亡。对骨组织来说,体外研究已经证实脂肪细胞分泌的因子对BMSCs可产生脂毒性,影响细胞分化功能[7]。脂肪细胞也可以对成骨细胞的功能和活性产生影响,其可被脂肪酸合成抑制剂阻断,且抑制脂肪酸合成可以促进成骨[8],说明脂肪酸可引起对成骨细胞的脂毒性。游离脂肪酸及其他脂毒性物质在细胞内的增多,会增加线粒体对脂肪酸的氧化或通过内质网应激等途径,影响细胞功能[9-10]。氧化应激介导的细胞功能异常是脂毒性一个重要的发病机制,如活性氧(reactive oxygen species,ROS)聚集可导致氧化应激,其具有双重作用,可氧化、破坏蛋白质、脂质、DNA,导致细胞功能改变,也可激活胞内多重适应性信号通路[11-12]。氧化应激可诱导成骨细胞凋亡已被广泛认可[13],且细胞的氧化应激以及ROS水平升高会促使 MSCs向成脂分化倾斜,导致成骨形成减弱及成骨数量减少[14]。Arai等[15]研究认为在氧化应激环境中成骨细胞分化下降与抗氧化酶系统表达上调和成骨基因表达的改变密切相关。由此可见,骨髓腔内脂肪细胞堆积通过氧化应激介导的脂毒性在抑制BMSCs成骨分化、成骨细胞活性,继而引起骨质疏松中扮演了重要角色。因此,通过探讨脂毒性通过氧化应激途径诱导骨质疏松的发病机制,将会给原发性骨质疏松的治疗研究带来新的切入点和靶点。
3 氧化应激介导的FoxO-β-catenin-PPARγ信号轴对BMSCs分化失衡的作用机制
经典 Wnt 信号通路在成骨细胞的分化、增殖中发挥重要作用,是成骨细胞的正向调节蛋白,而且还可以抑制破骨细胞的功能和分化[16]。这一通路主要由细胞外因子Wnt蛋白、糖原合成激酶3β(GSK3β)等介导,最终稳定的β-Catenin进入细胞核,调节靶基因的转录和表达,通过Runx2基因促进成骨的形成[17]。调控该途径中任何一个因子都可影响成骨细胞的功能与活性,从而对骨形成和骨代谢起着重要的调控作用[18]。现代研究也发现[19]随着年龄的增长,成骨细胞数量的减少与氧化应激增加抑制Wnt信号途径有关。
过氧化物酶体增殖活化受体(PPAR)是一类配体激活的核转录因子超家族成员,其中PPARγ是最具有脂肪细胞特异性、对脂肪细胞的分化起关键性作用的调节因子,还可抑制成骨分化[20]。临床观察也发现应用PPARγ激动剂治疗糖尿病,可引起糖尿病性骨质疏松[21]。研究表明[22],骨髓中PPARγ2随年龄的增加而增加,此外体内游离脂肪酸可促进PPARγ表达,诱导BMSC成脂分化,使骨髓腔中脂肪细胞堆积,抑制成骨分化[23],形成恶性循环; ROS 还可以增加脂氧合酶Alox15 的表达,继而反馈调节脂质氧化,后者又可以结合PPARγ,促使PPARγ与β-catenin 结合,进而降低β-catenin /TCF诱导的成骨细胞形成[24]。PPARγ在BMSCs成骨、成脂双向分化过程中起着“开关”的作用,表达升高,则抑制成骨分化,促进成脂分化[25]。
FoxO刺激机体表达与DNA修复和自由基清除有关的各种功能基因,并调节几种前凋亡基因,从而诱导异常或损伤细胞发生凋亡[26]。在哺乳动物细胞中,FoxO4、FoxO1和FoxO3a的过量可诱导细胞发育的停滞或凋亡[27-28],在过氧化氢诱导的氧化应激反应中,Foxo的活性与细胞内脂质氧化损伤和caspase-3活化有关[29]。进一步研究发现[30]细胞中敲除 FoxO1、FoxO3和FoxO4基因,骨骼中成骨细胞的数量会下降,氧化应激水平升高。其中FoxO1 转录因子主要参与细胞凋亡、应激、DNA 损伤/修复等生命过程,NaKae等[31]提出FoxO1在激素激活的信号通路与促进脂细胞分化的复合转录瀑布的整合方面起重要作用。Rached等[32]也发现FoxO1是成骨细胞氧化应激中调节骨重建一个关键调节因子; FoxO3[33]则在氧化应激反应时乙酰化作用增强。动物实验[34]表明,氧化应激是与年龄有关的骨质流失和骨强度下降的一个关键发病因素,FoxO是与β-catenin密切联系的转录因子,且拮抗Wnt信号的能力随着年龄的增加而增强。过多的ROS致使 Wnt/β-catenin信号通路TCF/LEF转录转向为 FoxO 转录,降低成骨,最终导致机体出现骨质疏松[35-36]。
脂质氧化能抑制经典的Wnt信号通路,引起成骨细胞数量与骨形成下降[19]。在脂质过载环境中,Wnt 信号通路与 FoxO信号通路及PPARγ信号通路间的交联作用影响骨质疏松的发生和进展。当ROS过多时,FoxO可竞争性的与β-catenin 结合,抑制Wnt信号通路中TCF/LEF 转录,降低成骨,出现骨质疏松[37],而β-catenin的过度表达,可以防止FoxO介导的基因转录;老化使骨髓MSCs的PPARγ2表达增加,同时脂肪酸氧化产物能与PPARγ2结合并激活PPARγ2,降解β-catenin蛋白,导致βcatenin/ TCF调节转录作用降低[35,38],Wnt也可通过PPARγ影响到脂肪细胞的分化[39]。FoxO -β-catenin-PPARγ交联轴(图1)所介导的氧化应激反应在骨骼中的作用逐渐受到研究者们的关注[19]。成鹏[40]等研究表明通过调节FoxO3a/Wnt2/β-catenin通路,抑制FoxO3a,可提高β-catenin蛋白表达水平,能够有效缓解氧化应激介导的去卵巢大鼠的骨质疏松。Philip等[41]也发现使用抗氧化剂n-乙酰半胱氨酸(NAC)可通过抑制Foxo的活性,解除对Wnt信号的拮抗作用来预防酒精诱导BMSCs分化失衡的作用。动物实验[42]表明敲除前成骨细胞的β-catenin基因,导致成年小鼠PPARγ2的表达增加,骨髓内脂肪堆积,骨量减少,而他汀类药物可以通过激活Wnt通路和抑制PPARγ通路,起到治疗脂代谢异常带来的骨质疏松和骨质流失的作用[43]。Gao[44]等研究发现,雌激素可通过经典Wnt通路抑制骨-脂肪形成,认为雌激素引起的Wnt通路活性下降是骨髓脂肪逐渐增多的重要原因。
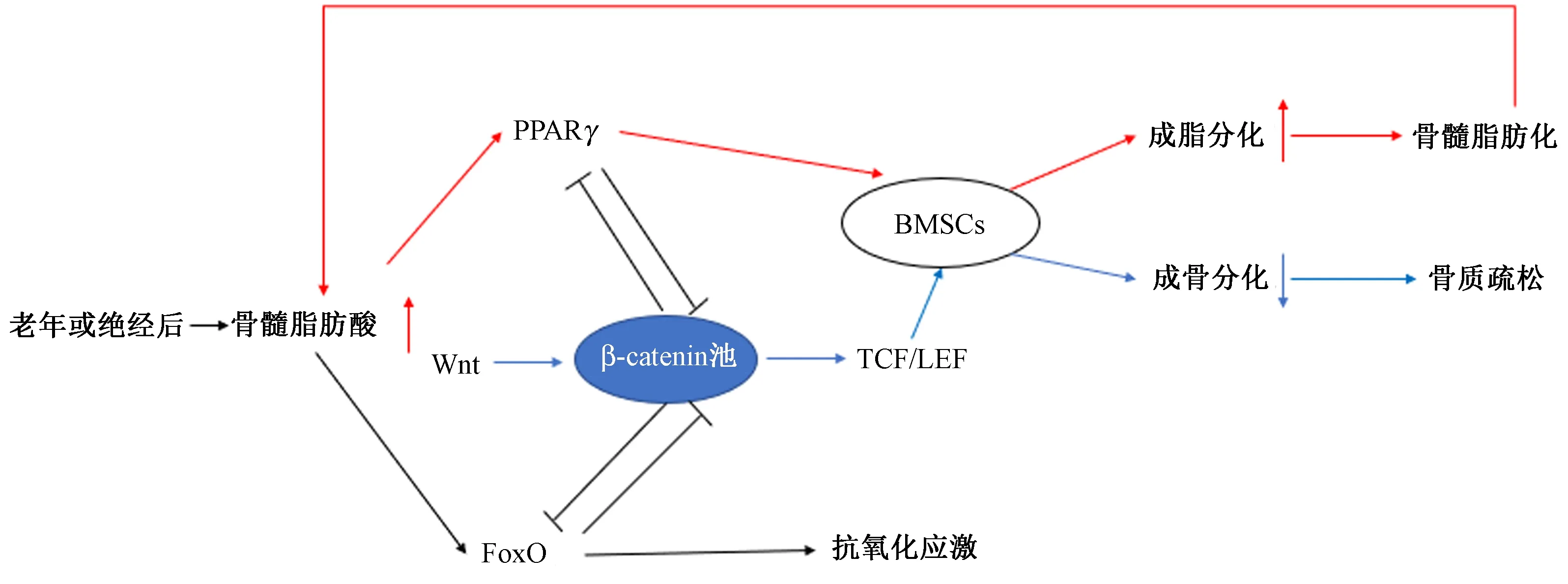
图1 FoxO-β-catenin-PPARγ信号交联图Fig.1 FoxO-β-catenin-PPARγ signal cross-linking diagram
综上所述,氧化应激介导的FoxO-β-catenin-PPARγ信号轴的平衡,可能在骨髓脂肪化微环境下,对BMSCs的成骨分化能力及成骨细胞活性起着重要作用,其中对β-Catenin浓度的调控处于中心地位,这或许可为原发性骨质疏松的治疗提供一个新的着眼点。
猜你喜欢
中国组织化学与细胞化学杂志(2022年3期)2022-09-18
天津医药(2021年6期)2021-12-08
口腔医学(2021年10期)2021-12-02
医学研究杂志(2021年10期)2021-11-26
中华骨与关节外科杂志(2021年12期)2021-08-31
天津医科大学学报(2021年3期)2021-07-21
天津医科大学学报(2021年3期)2021-07-21
山东医药(2020年36期)2020-12-31
医学新知(2019年4期)2020-01-02
文苑(2018年18期)2018-11-08
