
强直性肌营养不良症16例临床分析及文献复习
2019-08-28 06:39:58陈校文时宏娟花放
神经损伤与功能重建 2019年8期
陈校文,时宏娟,花放
强直性肌营养不良症(myotonic dystrophy/dystrophy myotonic,MD/DM)是一组以肌无力、肌强直和肌萎缩为特点的多系统受累的常染色体显性遗传病。国外流行病学报道发病率约3~15/10万,具有明显的种族及地域、性别差异[1]。本研究对16例DM患者的临床资料进行回顾性分析,并结合文献分析,以提高医师对该病的诊疗水平。
1 资料与方法
1.1 一般资料
纳入2011年1月10日至2018年5月8日我科收治的诊断为DM的患者16例的临床资料。
1.2 方法
收集16例患者的症状、体征,体格检查,实验室、影像学、电生理、病理检查及基因测序结果,分析其临床表现及特点。
1.3 统计学处理
采用SPSS 16.0软件处理数据。符合正态分布以及方差齐性的计量资料以(±s)表示,计数资料以率表示,进行描述性统计。
2 结果
2.1 一般情况
本组共纳入16例,其中男11例,女5例;年龄14~61岁,发病年龄12~51岁;病程1.5~14年。其中8例患者来自3个家系,每个家系至少有2名患者;7例为散发;1例自幼被收养,家族史不详。
2.2 临床表现
首发症状中7例表现为握拳或咀嚼后难以放松,遇冷风或冷水后加重;8例肌无力,1例为发作性头晕。病程中16例患者均出现肌肉无力,无力的肌肉分布以肢体远端为主,强直及无力及萎缩的肌肉分布见表1。强直主要表现为握拳或咀嚼后很难放松或面部表情肌僵硬,症状在重复动作后减轻,遇冷后加重;存在典型叩击性肌强直5例,表现为叩击双手大鱼际、舌肌、腓肠肌等出现肌肉紧张成肌球,难以放松。骨骼肌外的各个系统的受累情况见表2。
2.3 辅助检查
所有患者首次就诊时肌酶结果如下:肌酸激酶(creatine kinase,CK)80~568 U/L,平均253.19 U/L,(正常值20~180 U/L);肌酸磷酸激酶同工酶MB(creatine kinase isoenzyme-MB,CKMB)2.5~10.1 ng/mL,平均5.87 ng/mL(正常值0.0~3.61 ng/mL);乳酸脱氢酶(lactate dehydrogenase,LDH)140~653 U/L,平 均247.69 U/L(正常值110~240 U/L);谷丙转氨酶(alanine transaminase,ALT)11~58 U/L,平均25.75 U/L(正常值7~40 U/L);谷草转氨酶(aspartate transaminase,AST)14~51 U/L,平均29.44 U/L(正常值13~35 U/L)。
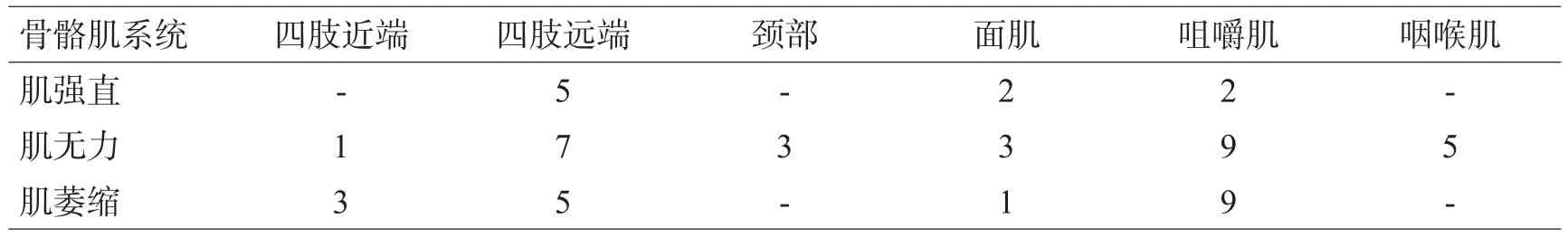
表1 本组患者强直、无力及萎缩的肌肉分布(例)
16例患者全部行肌电图检查,其中15例示四肢肌及面肌见有肌强直电位,11例肌源性损害伴强直电位,4例仅有强直电位,1例仅有肌源性损害无强直电位。6例患者行肌肉活检,提示:肌源性肌萎缩,符合强直性肌营养不良特点(图1)。
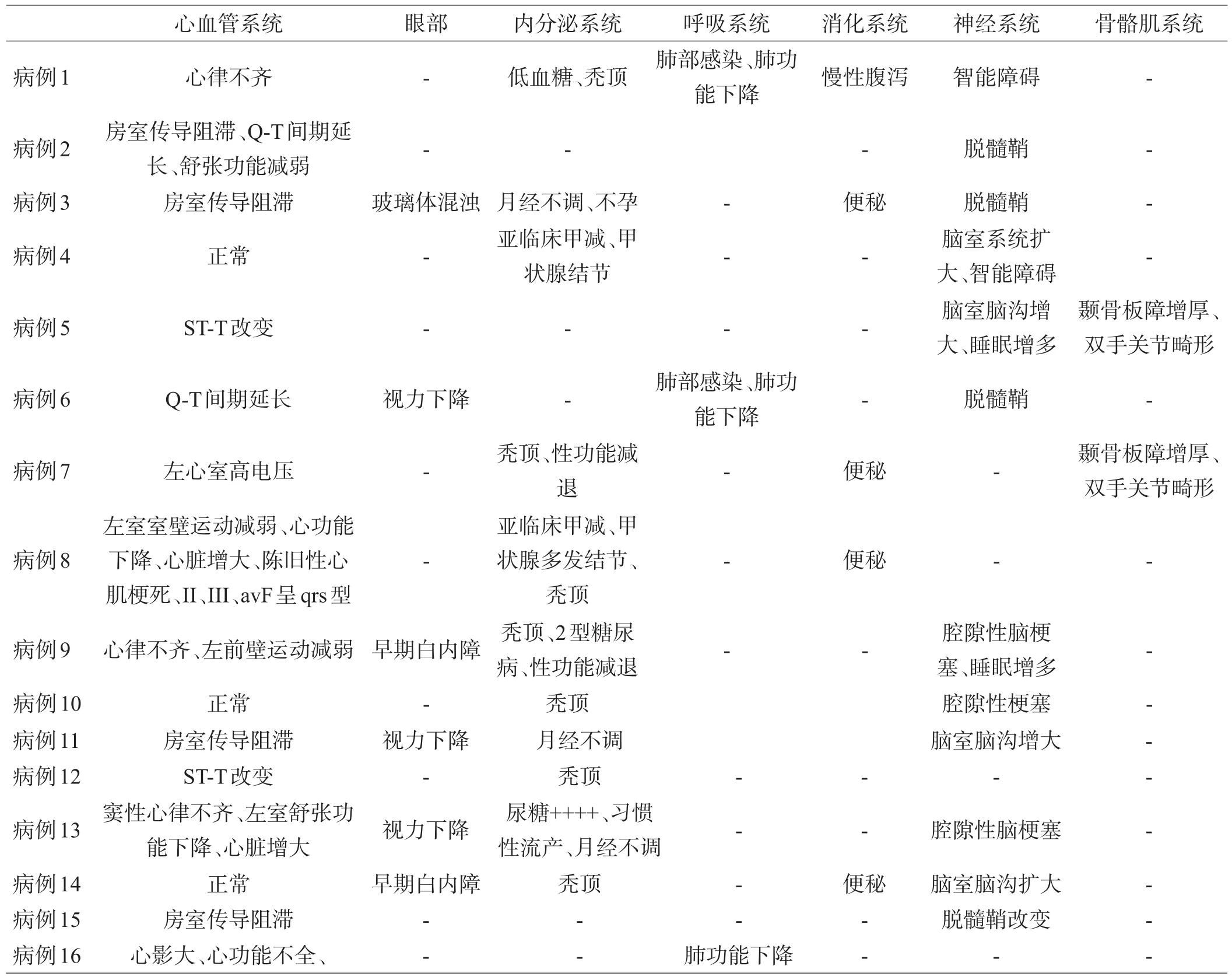
表2 本组患者骨骼肌外系统受累情况
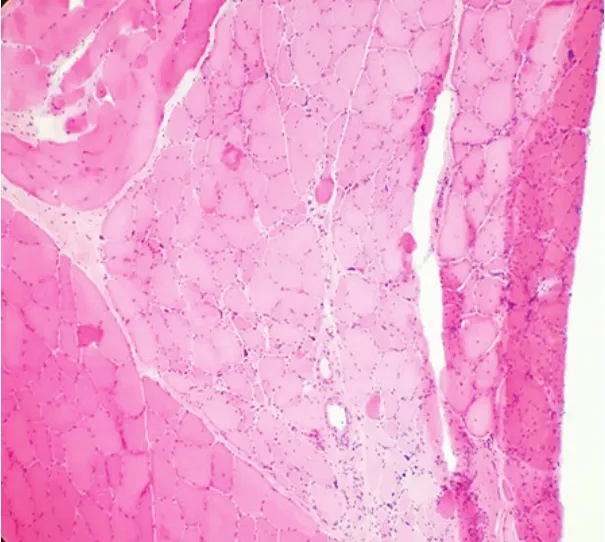
图1 患者肌肉活检HE染色
基本上所有患者都存在累及心脏系统且心脏隐患是DM患者最常见的猝死原因,本组患者中,窦性心律失常3例(18.8%),房室传导阻滞4例(25.0%),陈旧性心肌梗死1例(6.0%),单纯舒张功能减退2例(12.5%),T波改变2例(12.5%),全心脏增大3例(18.8%),Q-T间期延长2例(12.5%),左室肥大1例(6.0%),左室室壁运动减弱2例(12.5%)。
患者中7例行基因检查,外送至广州金域检验公司,均存在DMPK基因的3’UTR区的CTG重复数目,均>50次,属于全突变范围,提示符合DM1型基因突变特征。
3 讨论
DM是多系统受累的常染色体显性遗传病,分为前突变、晚发型、成年型(经典型)、少年型、婴儿型、先天型。除前突变外,其他类型均会使患者寿命缩短,先天型寿命最短。除前突变型外显率不确定可能存在变异之外,其余类型均为完全外显[2]。本组8例患者有阳性家族史,对本病诊断有重要提示,但因本病存在遗传早现[3],故家族史阴性不能排除本病诊断。
DM患者最突出的临床表现为肌肉强直、萎缩和无力,肌肉主动及被动收缩后出现放松困难,遇冷水或冷风后加重,而重复动作后可好转,称为“热身现象”[4]。随着病程进展,患者常出现全身多系统症状。最值得关注的就是心血管系统受累,有研究显示完全性房室传导阻滞及房颤是患者猝死的主要原因,心电图上的严重异常(定义为PR间期和/或QRS间期延长、II/III度心脏传导阻滞或非窦性心律)或房性快速心律失常预示猝死[5]。文献报道约70%患者出现各类型的心律失常,可见房室传导阻滞、窦性心律不齐、PR间期延长、QT间期延长、非窦性心律如房颤,部分患者出现心脏彩超提示结构性异常[6],如心脏增大、室壁运动不协调、二或三尖瓣脱垂甚至少见的如心肌病。本组心电图异常发生率为62.5%,以房室传导阻滞最为常见25.0%,但无法排除其他疾病引起的心脏异常。有报道称在年轻的患者中快速心律失常比房室传导阻滞更为常见[7]。本组心脏彩超提示结构异常31.25%,以全心增大及左室室壁运动减弱最为常见。呼吸系统受累时可出现肺功能下降、夜间呼吸睡眠暂停、肺部感染甚至呼吸衰竭等[8],本组观察到肺功能减低及肺部感染者。内分泌系统受累时可出现甲状腺,胰腺,下丘脑,性腺和甲状旁腺紊乱。在DM患者中,糖尿病发病率稍高于一般人群,可能与胰岛素受体的异常剪接而导致的胰岛素抵抗有关[9],疾病肌肉症状或心脏及肺部疾患使患者不能耐受运动导致肥胖也会增加糖尿病风险。本组68.75%的患者出现内分泌受累,主要累及胰腺、甲状腺及性腺,其中甲状腺及性腺受累最常见。因为并未做完整的性激素及甲状旁腺激素的检查,故可能结果存在误差。眼部受累时可表现为眼睑下垂、白内障、视力下降[10],也可发生玻璃体混浊、结膜炎、角膜炎等眼科疾病。胃肠道可以出现与肠易激综合征相似症状,如腹痛、吞咽困难、呕吐、慢性或发作性腹泻、进食时咳嗽、和肛门失禁等[11]。神经系统受累及时可出现嗜睡、认知和智力缺陷,但症状表现无特异性,起病隐匿,与发病年龄及病程进展有关。头颅MRI白质病变(脱髓鞘)、脑萎缩等最常见[12,13]。较少见的表现如骨骼系统可出现蝶鞍小、脊柱侧弯、多种骨变形,本组患者中亦可见2例掌指关节畸形合并颞骨板障增厚。平滑肌受累时可出现如食管扩张、巨结肠、胃肠蠕动功能下降表现,胆囊可因为排空不佳导致胆囊结石。典型的DM诊断结合典型的多系统受累临床表现、肌电图提示强直电位。肌活检见肌纤维大小明显不等,内核纤维增多及选择性I型纤维萎缩为主。基因检测提示位于19q13.2-13.3上DMPK3‘端非编码区CTG三核苷酸重复序列的异常扩增>50可确诊。
目前DM尚无特效治疗办法,基因敲除在动物实验取得一定成果。目前临床上所采用的主要是对症治疗。肌肉强直症者可用苯妥英钠、普鲁卡因胺等改善强直症状,但在存在房室传导阻滞的患者,普鲁卡因胺应慎用;除骨骼肌症状外,还应关注其骨骼肌外症状,尤其是心肺功能,文献报道心肺疾病造成强直性肌营养不良患者死亡率的70%[14]。提示在确诊的患者及无症状的前突变者的后代中,及时发现心脏及呼吸异常至关重要。凡DM患者无论有无心脏异常均推荐每年心电图检查,必要时24 h心电图检查,应尽量避免应用可能导致心脏传导阻滞及血流动力学不稳的药物;出现III度或高度房室传导阻滞时推荐起搏器植入,房颤推荐及时药物治疗;心衰予以对症处理;必要时心内科专科治疗[14]。消化系统有腹胀消化不良时可予以饮食调整及多潘立酮等对症处理;内分泌系统出现糖尿病、钙缺乏、甲状腺功能减退时分别予以胰岛素、维生素D3补充及甲状腺激素治疗;但DM患者任何手术均需在评价肺功能基础上完善。10%的风险是由于全身麻醉,包括麻醉剂的长期呼吸抑制和术后肺炎,尤其是胆囊切除术后,强调有必要进行DM患者的围手术期处理[15]。因DM属于常染色体显性遗传疾病,产前诊断(羊膜腔穿刺、超声引导下经阴道或经腹绒毛取样)可为后代提供选择性终止妊娠的选择。有条件时可选择植入前遗传学诊断[16]。
DM起病形式多样,既往有胸痛、晕厥及急性冠脉综合征起病的DM病例报道[17,18],随病程进展可累及多个系统,病情严重程度相差很大,甚至在一个家族中可见无症状杂合子到病情严重的婴幼儿。尤其是当肌肉症状不明显而以骨骼肌外症状突出时,很有可能被忽视,导致在疾病的早期出现误诊、漏诊。神经科医师应提高对此病的认识,及早的识别骨骼肌外尤其是心脏受累症状,并积极治疗,提高患者生存质量。
