赖氨酸甲基转移酶SETD6在肿瘤发生发展中的研究进展
2019-08-20 01:43周慧,陈攀,许静,陈孝,陈杰
中国药理学通报 2019年8期
周 慧,陈 攀,许 静,陈 孝,陈 杰
(1. 中山大学附属第一医院药学部,广东 广州 510080;2. 南方医科大学附属皮肤病医院药剂科,广东 广州 510091)
蛋白质转录翻译后可进行甲基化修饰,甲基化是一种普遍的修饰形式,调控着蛋白质的功能。能够甲基化赖氨酸残基的这类含有SET保守结构域的蛋白叫做赖氨酸甲基转移酶[1]。赖氨酸甲基转移酶SET家族包含SETD1~SETD9、G9a、MLL、SUV39H1等50多种人源相关蛋白。其著名家族成员SETD7(SET domain containing 7)能够甲基化p53蛋白上第372位赖氨酸,形成一甲基化物K372me1,激活p53蛋白的转录活性,从而抑制肿瘤细胞的增殖[2]。近几年,人们发现家族中另一重要成员赖氨酸甲基转移酶SETD6(suvar3-9, enhancer of zeste, trithorax domain containing 6)可参与到多种重要的信号通路中,通过修饰相关蛋白结构和影响蛋白活性,在肿瘤细胞中发挥作用,且与肿瘤的发生、发展息息相关[3]。因此,本文综述了SETD6在肿瘤细胞中的功能、相关的分子基础及信号通路等研究进展,并进一步探讨该蛋白成为治疗肿瘤新靶点的应用前景。
1 SETD6的蛋白结构和功能
SETD6是SET蛋白家族中的一员,同属于赖氨酸甲基转移酶。其蛋白编码基因位于人体第16号染色体,包含47个外显子,编码2 034个氨基酸残基,蛋白质分子量为53 ku。SETD6是个核质蛋白,由i-SET(是指插入约125个氨基酸的中间SET区域)和C-SET(是指C端SET,是编码螺旋结构的主要基因序列)编码的螺旋结构在底部相接,使整个蛋白呈现V字样结构[4],该V字结构是SETD6蛋白发挥功能的结构基础。整个蛋白由1个含有SET固定结构的催化域、1个底物结合域,以及两个假定的LxxLL核受体相互作用基序组成。第一个LxxLL基序深埋于含有SET固定结构的催化域中,第二个LxxLL基序位于SETD6蛋白表面的羧基端,该基序由于空间位置结构的优势成为最有可能与核蛋白受体相互结合的位点[5]。赖氨酸甲基转移酶的功能最早被认为是对组蛋白转录后的甲基化修饰,例如甲基化组蛋白H2A的变异体H2A.Z第4位和第7位的赖氨酸,形成甲基化物H2AZK4mel和H2AZK7me1,两种甲基化物有协同作用,可维持鼠胚胎干细胞的自我更新功能,因此,甲基化物H2AZK7mel被认为是细胞分化的标志[6]。近年来,人们发现非组蛋白上也能发生甲基化修饰,例如SETD6可甲基化RelA(p65)蛋白第310位赖氨酸,一定程度上阻碍了p65的磷酸化,最终下调NF-κB通路相关蛋白的表达,抑制该通路介导的炎症反应[7]。
SETD6不仅是甲基化修饰的功能蛋白,还是核受体信号共激活因子,它一方面可以通过甲基化修饰调控蛋白底物活性,进而影响相关通路上的靶基因转录活动;另一方面还可以与核受体共激活因子作用,激发转录活性,最终影响细胞的增殖、分化、炎症反应,见Tab 1。
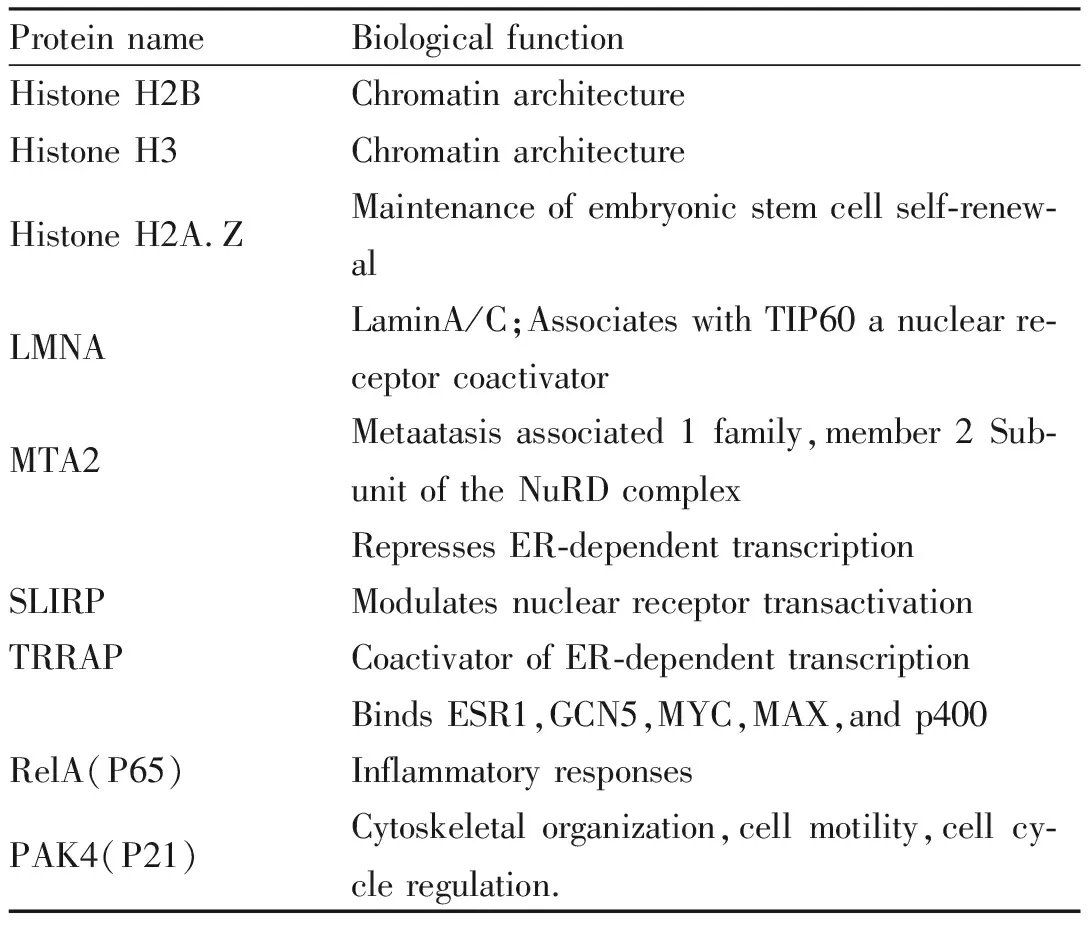
Tab 1 Nuclear receptor signaling factors, some mainproteins associated with SETD6 and their biological functions
2 SETD6在肿瘤细胞中的作用
2.1 SETD6与结肠癌Martin等[8]对X型家族性结肠癌患者的外显子进行测序时发现,3位患者的肿瘤组织和正常细胞中均出现了罕见的SETD6基因编码突变,3位健康成员则没有该突变。SETD6编码基因的突变不仅损害了SETD6正常功能,并且使突变者具有更高的结肠癌患癌风险。突变是由SETD6正常蛋白的264位和265位甲硫氨酸和丙氨酸被异亮氨酸(Ile264)和甘氨酸(Gly265)移位代替导致了过早的转录终止,突变的等位基因编码并翻译出只含有N端的SETD6-N。这种突变使得SETD6蛋白C-端缺失,即便SETD6基因编码的蛋白保存了SET结构域,但甲基转移酶活性缺失。突变的SETD6-N和野生型SETD6(wild type SETD6,SETD6-WT)在结肠癌细胞的染色质中竞争结合底物RelA,SETD6-WT与RelA结合后将其甲基化形成RelAK310me1,甲基化物RelAK310me1反过来又可以抑制SETD6-N的过表达,两者呈现剂量依赖的关系。
2.2 SETD6与乳腺癌沉默SETD6的MCF7乳腺癌细胞系的生长曲线出现明显地增殖缺陷,G1时期的细胞数量占比从2%升至20%~35%[5];其他乳腺癌细胞系,例如MDA-MB231细胞中,下调SETD6蛋白表达后细胞周期抑制剂CDKN1A的表达量升高为正常值的6倍[5]。这些数据表明SETD6基因沉默后可抑制肿瘤细胞增殖,扰乱肿瘤细胞周期进程,诱发凋亡。Daniel等[5]证实SETD6蛋白调控乳腺癌细胞的增殖机制与核激素受体信号相关,例如SETD6蛋白能与雌激素受体α(estrogen receptor α,ERα)结合并提高雌二醇的表达。过去的研究已知雌激素受体α(estrogen receptor α,ERα)与雌激素结合可促使乳腺细胞增殖[9],对于ERα基因阳性的乳腺癌患者采用抗雌激素疗法能够控制肿瘤细胞的增殖活动[10]。然而该方法对于ERα基因阴性的突变患者无效,并且较大比例的ERα基因阳性患者出现药物抵抗,最新研究发现SETD6蛋白下调可使乳腺癌细胞增殖能力降低,且该作用独立于ERα基因,这使得SETD6在调控乳腺癌增殖方面不再受基因型的影响。
除此之外,SETD6蛋白还被发现是雌激素响应基因的共激活因子,沉默SETD6基因的MDA MB231 (ER-, PGR-,p53mut),MCF7 (ER+,PGR+, p53wt)以及T47D (ER+, PGR+,p53mut) 细胞系中孕酮受体基因(progesterone receptor gene,PGR)、TFF1以及其他雌激素反应基因的表达量都大幅度减少。这些信息表明,SETD6基因的抑制或沉默将有潜力成为控制乳腺癌进展的又一手段。
2.3 SETD6与膀胱癌Mukherjee等[11]发现,膀胱癌患者的癌组织中SETD6蛋白水平明显高于正常组织,而正常组织中SETD6蛋白几乎不表达。同样地,在膀胱癌细胞株T24中,SETD6 mRNA和蛋白水平也明显高于正常细胞表达,提示SETD6蛋白与膀胱癌有密切关系。上调SETD6的膀胱癌T24细胞中,生长曲线和克隆形成能力增强,沉默SETD6基因后,这两项参数均下降。在低内源性表达SETD6蛋白的SVHUC1膀胱癌细胞系中,外源性补充SETD6蛋白后,明显增加了细胞克隆形成,并且使增殖期细胞百分比升高。在高内源性表达SETD6蛋白的膀胱癌细胞株RT4和UMUC3中,利用siRNA下调SETD6蛋白表达后,细胞生长受到抑制,细胞死亡百分比升高。这些结果都表明,SETD6蛋白能够促进肿瘤细胞生长增殖,提高细胞克隆能力,维持细胞的生长活力。除此之外,研究还发现,SETD6蛋白在皮肤癌、胰腺癌、前列腺癌中表达明显高于正常组织。这些结果表明,SETD6蛋白与多种肿瘤具有相关性并起到促进其增殖的作用。
3 SETD6参与肿瘤细胞增殖存活的分子机制
3.1 SETD6甲基化PAK4调控Wnt通路,促进肿瘤细胞增殖和迁移p21活化激酶4 (p21-activated kinase 4,PAK4)属于丝氨酸/苏氨酸蛋白激酶家族中的成员,具有调节细胞骨架、细胞增殖、凋亡、迁移等功能[12]。研究发现,下调PAK4蛋白表达之后,非小细胞肺癌细胞株A549和NCI-H520的侵袭、迁移能力明显下降,而上调PAK4蛋白表达在多种肿瘤中具有促进细胞迁移的作用[13]。PAK4蛋白可将β-catenin蛋白磷酸化,磷酸化的β-catenin与axin/APC、GSK-3β复合物分离,并移位到细胞核与TCF/LEF结合,触发Wnt信号通路,从而促进细胞增殖、迁移、耐药等特性[14]。SETD6可在核染色质内结合PAK4并将其甲基化,PAK4在细胞核与细胞质之间的循环动态调节了由β-catenin介导的Wnt信号通路的活动[15]。在沉默SETD6基因的TMDA-MB-231细胞系中,β-catenin的多种下游目标基因表达水平急剧下降,这一现象表明SETD6在Wnt/β-catenin转录过程中有一定的调控作用,被SETD6蛋白甲基化的PAK4蛋白从核质转位到胞质,将β-catenin第675位的丝氨酸磷酸化,从而稳定地促进转录活动。综上所述,SETD6蛋白在PAK4/β-catenin信号通路中是一个关键的调控因子,沉默SETD6基因后能够明显降低多个细胞系中PAK4/β-catenin通路的下游基因的表达。而PAK4/β-catenin通路在非小细胞肺癌等多种肿瘤中具有重要作用,例如Akiri等[16]的研究发现,50%人类NSCLC患者的细胞株中有Wnt通路失调。Xu等[17]在NSCLC患者肺癌组织免疫组化的研究结果显示,36.6%的胞内Wnt1表达上调,且与β-catenin的表达上调有明显相关性。滕颖[18]的研究发现,Wnt/β-catenin信号通路激活的肺腺癌A549细胞的克隆形成、迁移、耐药能力等特性增强;Wnt/β-catenin信号通路抑制的A549细胞内更多的细胞发生G0~G1期阻滞,细胞生长能力下降,同时上述几个细胞特性受到抑制。因此,SETD6蛋白在调控肿瘤增殖迁移的方面具有一定的研究潜力。
3.2 SETD6通过抑制DJ-1蛋白的活性下调氧化应激通路DJ-1蛋白在控制活性氧自由基水平以及降低氧化应激引起的细胞凋亡方面,起着重要的作用[19]。Chen等[20]在人类红白血病细胞K562、人胚胎肾293T细胞和人乳腺癌MDA-MB-23细胞模型中证实了内源性的SETD6与DJ-1能够相互结合,使得从胞质中转位到核质中的DJ-1蛋白呈现SETD6/DJ-1复合物形式,DJ-1蛋白无法发挥活性,由它所激活的Nrf2-ARE通路相关下游基因表达下调,细胞氧化应激反应减弱,SETD6蛋白抑制DJ-1蛋白活性的作用并非通过甲基化修饰。
越来越多的证据表明,DJ-1蛋白在肝细胞癌、乳腺癌、胰腺导管腺癌、非小细胞肺癌高表达,而在下调DJ-1蛋白的胰腺导管癌细胞株中,细胞在体外迁移和体内侵袭的潜力均下降。DJ-1不仅在细胞氧化应激方面起到正向激活的作用,而且在肿瘤细胞增殖和转移的正向调控蛋白。现有的文献研究资料仅阐述了SETD6蛋白与DJ-1蛋白相互作用后下调细胞氧化应激反应的现象和原理,对于两种蛋白相互结合在肿瘤细胞中的作用却仍处于空白。
3.3 SETD6与NF-κB通路的关系NF-κB是炎症反应的关键诱导因子,与人类众多疾病,如肿瘤、免疫失调和自身免疫疾病相关。NF-κB的激活不仅能促进肿瘤细胞增殖,抑制细胞凋亡,还能促进血管生成,诱发上皮间质转化,从而使肿瘤细胞向远处转移。NF-κB家族的主成员有RelA(p65)、NF-κB1(p50)、NF-κB2(p52)等。虽然正常状态的p65蛋白存在于细胞质中,并与IκB蛋白家族的成员结合而处于无活动状态,但是在未刺激条件下的人类胚胎肾293T细胞和人骨肉瘤U2OS细胞还发现核质中存在RelA的一甲基化物RelAK310mel,这一现象表明了还有一定数量的RelA处于未结合状态,并转位到核质被SETD6甲基化[7]。Levy等[7]发现,SETD6蛋白的存在与NF-κB介导的炎症之间呈现负相关,表明SETD6甲基化p65蛋白后,由NF-κB介导的炎症反应减弱。但是另一研究发现[11],膀胱癌细胞中SETD6过表达时,反而出现了p65 mRNA和蛋白水平的升高,同时p65抑制配体IκBα的mRNA和蛋白水平下降。SETD6蛋白表达上调的情况下,检测到p65和磷酸化p65两种蛋白的表达水平升高。在这里SETD6蛋白又变成了NF-κB的诱导剂,正向影响着p65的蛋白表达,提高通路下游基因p21、NQO1、CCL2及MDM2的表达,对肿瘤细胞增殖起促进作用。
现有研究资料显示着两种相悖现象,这可能与SETD6蛋白结构相关,当p65蛋白第311位丝氨酸未磷酸化激活时,SETD6可在其相邻位(第310位赖氨酸)甲基化修饰,甲基化的p65蛋白磷酸化过程受阻,其激活的各种转录过程丧失;但是很大部分的p65经磷酸化活化后才入核,此时甲基化作用受阻,SETD6将无法下调其介导通路的各种活性。随后人们还发现,p65甲基化物可被另一甲基化酶修饰酶GLP(G9A样蛋白)上的ankryin重复序列识别,稳定GLP对组蛋白H3的第9位赖氨酸(H3K9)的甲基化修饰,并且抑制相关基因的表达[5]。这也许印证了SETD6为何在不同细胞中截然相反的结果,SETD6发挥作用的途径不仅只有甲基化修饰,还可能与蛋白结构、蛋白结合活性等机制相关,这一领域有待科研工作者更多的探索。
4 总结和展望
综上所述,SETD6作为赖氨酸甲基转移酶可以将多种蛋白甲基化修饰,通过改变蛋白结构特性,使得蛋白活性发生改变或促使蛋白转位,进而调节目标蛋白所在通路的靶基因的转录。例如,SETD6通过甲基化PAK4蛋白,使其从细胞核转移到细胞质,磷酸化β-catenin蛋白,激活Wnt通路,促进肿瘤细胞的迁移;SETD6还可以甲基化组蛋白变异体H2A.Z,进而稳定胚胎干细胞的自我更新功能。SETD6研究最多的通路集中在通过甲基化p65蛋白,抑制NF-κB通路介导的炎症反应,人们将SETD6认为是炎症反应的负调节器。但是在不同的细胞状态时,SETD6发挥的功能不尽然相同,在膀胱癌细胞中就证实SETD6不仅可以上调NF-κB蛋白表达,还能上调p65及磷酸化p65的蛋白表达水平,这些现象直接导致肿瘤细胞的生存和克隆能力增强。无论是NF-κB或Wnt/β-catenin都与肿瘤的发生、增殖、转移等密切相关,因此,SETD6很有可能在肿瘤中起到重要作用,并且是一个促进肿瘤增殖、存活的关键蛋白。
猜你喜欢
热带作物学报(2022年7期)2022-08-06
保健与生活(2022年11期)2022-06-09
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
湖南饲料(2021年4期)2021-10-13
保健与生活(2021年5期)2021-04-12
陕西农业科学(2019年4期)2019-05-13
时尚育儿(2018年8期)2018-05-14
分析化学(2017年12期)2017-12-25
安徽医科大学学报(2015年9期)2015-12-16