
四倍体野生种花生A.monticola全基因组SSR的开发与特征分析
2019-08-13 07:58王玉龙黄冰艳王思雨杜培齐飞艳房元瑾孙子淇郑峥董文召张新友
中国农业科学 2019年15期
王玉龙,黄冰艳,王思雨,3,杜培,齐飞艳,房元瑾,孙子淇,郑峥,董文召,张新友
四倍体野生种花生全基因组SSR的开发与特征分析
王玉龙1,2,黄冰艳2,王思雨2,3,杜培2,齐飞艳2,房元瑾2,孙子淇2,郑峥2,董文召2,张新友1,2
(1河南科技大学农学院,河南洛阳 471023;2河南省农业科学院经济作物研究所/农业部黄淮海油料作物重点实验室/河南省油料作物遗传改良重点实验室,郑州 450002;3郑州大学生命科学学院,郑州 450001)
【】通过对四倍体野生种花生(AABB,2n = 4x = 40)全基因组SSR位点搜索,研究其全基因组SSR分布特征及规律,开发并验证其全基因组SSR引物,为花生属植物遗传进化分析及重要性状分子标记开发提供依据。【】在华大基因GigaScience数据库下载全基因组序列,并利用生物信息学软件MISA进行SSR位点搜索,Primer 3进行引物设计,通过电子PCR进行单位点SSR分析,并随机合成100对SSR引物验证通用性。【】SSR在四倍体野生种花生基因组上共搜索到SSR位点676 878个,平均每3.8 kb就会出现一个SSR,分布于5 127条scaffold中,单核苷酸至六核苷酸均有分布,且数量上差异较大,以单碱基、二碱基、三碱基为主,三者占SSR总数的94.28%,其中单碱基重复数量最多,占46.71%,密度最高;六核苷酸重复数目最少,分布最稀疏。大多数SSR分布在基因间区,基因区SSR多分布于内含子区域;全基因组共鉴定出395个不同的重复基元,其中A亚基因组342种,B亚基因组356种;A/T是最丰富的重复基元;在1—6个核苷酸的重复基元中,数量最多的依次是A/T、AT/AT、AAT/ATT,AAAT/ATTT、AAAAT/ATTTT、AAAAAT/ATTTTT;整体来看,重复基元的重复次数多集中在50次以内,不同类型的motif的重复次数差异很大;同一种类型重复基元的SSR位点,随着motif重复次数增加,SSR的数量逐渐降低;B03染色体上SSR数量最多,A08染色体中SSR密度最高。全基因组SSR比、基因组SSR数量多,密度也更高,单核苷酸重复最丰富,2个野生种二核苷酸数量最多。共设计出SSR引物192 303对,单位点SSR标记检出率50.35%,单点SSR标记在基因组上的分布呈现两端密集,中间稀疏的特点;随机合成的100对引物中,90对能在中扩增出稳定清晰的条带,且在4份不同的花生基因组DNA中扩增目的条带表现出不同的特点。【】基因组内SSR种类和数量丰富单核苷酸至六核苷酸均有分布,单核苷酸重复基元数量最多,且最密,六核苷酸重复基元数量最少,出现频率最低,不同重复基元频数高低与核苷酸数量没有严格相关性,SSR多分布在基因间区,基因区内含子区域SSR数量最多;A、B亚基因组具有其各自特异的重复基元类型;单个类型重复基元数量最多的均为AT富集的重复基元,而GC富集的重复基元相对较少;同一种类型重复基元的SSR位点,随着motif重复次数增加,SSR的数量逐渐降低;全基因组SSR较2个二倍体野生种数量更多,密度也更高且重复基元分布规律不同;经过初步验证,开发的SSR引物在4份花生材料中表现出部分通用性。
花生;;全基因组序列;SSR位点;基序
0 引言
【研究意义】花生是世界重要的油料作物和经济作物,是人类食用植物油和植物蛋白的重要来源,主要种植在亚洲、非洲和美洲等100多个国家和地区。中国花生年产量居世界首位,花生在国民经济中具有重要地位。栽培花生的2个二倍体祖先野生种(AA)和(BB)[1]及其近缘四倍体野生种(AABB)[2]全基因组序列已公布,对其基因组序列信息的深入挖掘和有效利用,开展全基因组范围内SSR(simple sequence repeats)的研究,探究其分布规律,并进行引物开发,将对花生属植物遗传进化分析及分子标记开发起到重要作用。【前人研究进展】SSR是指由1—6个核苷酸组成的不同类型基序多次重复而形成的相对较短的DNA序列[3]。在DNA复制、修复以及重组过程中DNA聚合酶滑动错配及后续的错误产生[4],其重复次数高度可变且侧翼序列相对保守,广泛分布于原核生物和真核生物基因组中[5-7]。研究表明SSR能够影响基因调控、转录、进化、蛋白质功能、基因组结构、DNA复制以及细胞循环等[8-9]。SSR分子标记是建立在PCR基础上的DNA分子标记,与其他分子标记相比,SSR具有多态性高、可重复性好、操作简便、及共显性等优点,是目前最常用的分子标记技术之一。SSR分子标记在花生属植物中也得到了广泛的应用,包括F1真杂种的鉴定[10]、指纹图谱的构建[11-13]、品种遗传多样性分析[14-18]、连锁图谱构建及QTL定位[19-23]等。目前,花生SSR分子标记的开发主要通过以下6种方式:(1)通过构建基因组文库的方式(包括基因组文库富集法和筛选基因组文库法)进行SSR标记开发[24-32];(2)基于公共数据库及自己开发的EST(expressed sequence tags)序列进行SSR标记的开发[33-40];(3)通过同源转移法进行SSR标记的开发,利用近缘物种开发的SSR引物应用到花生上[41];(4)基于构建BAC文库进行SSR标记开发[42];(5)基于转录组数据的SSR标记开发[43-44];(6)基于全基因组序列的SSR标记开发[45-46]。以上研究中花生SSR标记的开发多以栽培种为研究对象,对野生种花生的研究相对较少。2012年,Zhao等[47]整合了栽培花生和野生花生中SSR标记的总数为9 274个;Kazusa Marker数据库[48]收集了9 893个花生SSR标记,花生SSR标记逐渐丰富。【本研究切入点】花生SSR标记开发虽取得一些进展,但由于花生基因组庞大且复杂,遗传基础狭窄,SSR标记多态性较低,需要开发更多的SSR标记,以适应花生遗传研究。传统的SSR标记开发费时费力,目前,高通量测序技术高速发展,植物全基因组测序成本逐渐降低,为SSR序列的开发提供了极大便利。在拟南芥[49]、水稻[49]、黄瓜[50]、烟草[51]、短柄草[52]、葡萄[53]等植物中,已有大量的文献报道其基因组序列中SSR的信息,Yu等[54]开发了用于研究植物(包含110种)中微卫星DNA和SSR标记的数据库。栽培花生二倍体野生种祖先和四倍体野生种全基因组的测序完成,使花生的SSR高效开发成为可能。Zhao等[45]对栽培花生的2个二倍体野生种祖先首先进行了全基因组序列SSR分析,探究了其分布规律,共开发了204 439个SSR标记。但目前对四倍体野生种全基因组范围内SSR的研究,鲜见报道。【拟解决的关键问题】本研究采用生物信息学方法在全基因组序列中开展SSR位点分析,对基因组中不同区域及各条染色体的SSR位点进行统计,探究其分布规律,并利用搜索的SSR两端的保守序列进行引物设计继而开发出单一位点SSR分子标记。为进一步研究花生基因组的进化,真假杂种F1的鉴定,品种多样性分析,遗传图谱的构建等提供便利和理论依据。
1 材料与方法
1.1 植物材料
选用花生区组的4个不同种的花生材料为基因组DNA模板进行SSR引物的扩增验证。2个二倍体野生种花生分别为栽培花生的祖先种、1个四倍体野生种和1个栽培花生品种(表1)。
表1 供试材料的基本信息
1.2 基因组序列
全基因组序列及基因注释文件下载自华大基因GigaScience数据库,和基因组序列来自花生基因组数据库(PeanutBase)。基因组大小及组装水平见电子附表1。
1.3 基因组序列中SSR的搜索
利用生物信息学SSR搜索工具MISA(http://pgrc. ipk-gatersleben.de/misa/)进行全基因组中满足定义的SSR位点检索。为方便与栽培花生的2个野生种祖先和全基因SSR位点数量与分布特征比较,查找标准与Zhao等[45]所用标准保持一致:单碱基重复≥12 bp(基序M=1;重复次数N≥12);2碱基重复≥12 bp(M=2;N≥6);3碱基重复≥15 bp(M=3;N≥5);4 碱基重复≥16 bp(M=4;N≥4);5 碱基重复≥15 bp(M=5;N≥3);6 碱基重复≥18 bp(M=6;N≥3)。混合微卫星中2个SSR距离小于100 bp。利用Excel 2016、Perl脚本和R语言对所得数据进行统计分析与绘图。
1.4 A.monticola全基因组SSR引物设计
根据鉴定的四倍体野生种花生全基因组SSR位点侧翼的保守序列,利用引物设计工具Primer 3(Linux版)进行引物设计,引物长度18—23 bp,退火温度50—65℃,GC含量为40%—60%,扩增产物长度为100—300 bp。
1.5 电子PCR进行单位点SSR标记分析
利用获得的SSR引物在全基因组进行电子PCR扩增及电子定位,去除有多处扩增的引物,保证引物扩增的特异性。
1.6 SSR引物通用性验证
随机选取100对引物在通用生物系统(安徽)有限公司进行引物合成(电子附表2)。采用CTAB法提取4种供试材料的基因组DNA。PCR总反应体系为10 μL,包括20 ng·μL-1DNA样品2 μL、10×PCR Reaction Buffer(Mg2+plus)1 μL、dNTP Mixture 0.8 μL、ddH2O 5.75 μL、TaKaRa Taq 0.05 μL(5 U·μL-1)、正反向引物各0.2 μL。PCR反应程序为94℃5 min;94℃30 s,55—51℃45 s,72℃45 s,30个循环;72℃7 min,4℃保存。
完成PCR循环48 h内取1.5 μL产物在8%非变性聚丙烯酰胺凝胶上恒电压220 V电泳1 h 20 min,用NaOH银染方法进行染色显影。显影完成之后,拍照保存。
2 结果
2.1 A.monticola全基因组SSR位点数量和分布
利用MISA软件,在全长分别为1 084 261 490、1 353 826 449和2 618 653 824 bp的花生二倍体祖先种、和四倍体野生种基因组内分别搜寻到满足条件的SSR位点,基因组中共搜索到SSR位点676 878个,二倍体祖先种分别搜索到135 529和199 957个,与Zhao等[45]的结果一致。基因组检测到的SSR总长16 896 431 bp,占基因组的0.6452%,分布于5 127条scaffold序列中,平均3.8 kb出现一个SSR序列。A亚基因组平均4 kb出现一个SSR,B亚基因组平均3.9 kb出现一个SSR。SSR在四倍体野生种花生基因组上种类丰富,单核苷酸至六核苷酸均有分布,且数量上差异较大(表2)。在全部的6种基序(单、二、三、四、五、六碱基)类型中,单一重复类型(单个重复基元)SSR有538 886个(79.61%),而复合型(多个重复基元)SSR则有137 992个(20.39%)。单一类型重复基元数量最多的是单碱基重复,达到316 197个,其次是二碱基重复,三碱基重复和五碱基重复。基因组SSR位点主要集中在一、二、三碱基基序类型上,且三者约占总SSR位点数目的94.28%,分别为46.71%、30.76%和16.81%。五碱基基序类型的SSR位点所占的比例高于四、六碱基基序类型,六碱基基序类型在6种基序类型中出现频率最低,不同重复基元频数高低与核苷酸数量没有严格相关性(图1)。A、B染色体组各类型重复基元丰富度表现与全基因组相一致,且B基因组SSR位点数量更多。
SSR位点在基因组上的数量分布如表3所示,基因间区SSR数量最多,58 755个SSR位点分布在基因区。在基因区,内含子区域SSR数量最多,其次是CDS区域、UTR区域。
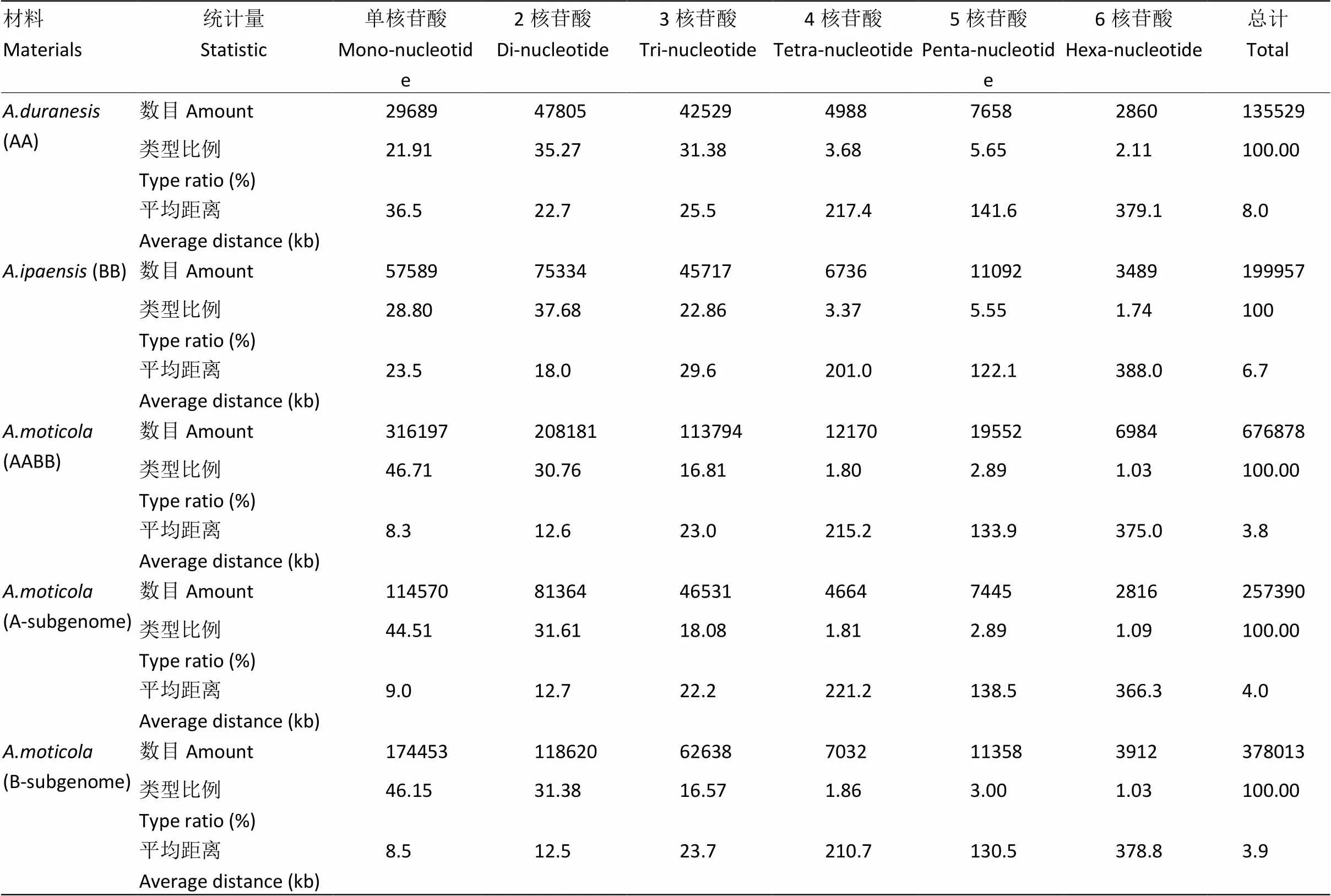
表2 A. monticola、A. duranensis和A.ipaensis基因组SSR位点信息
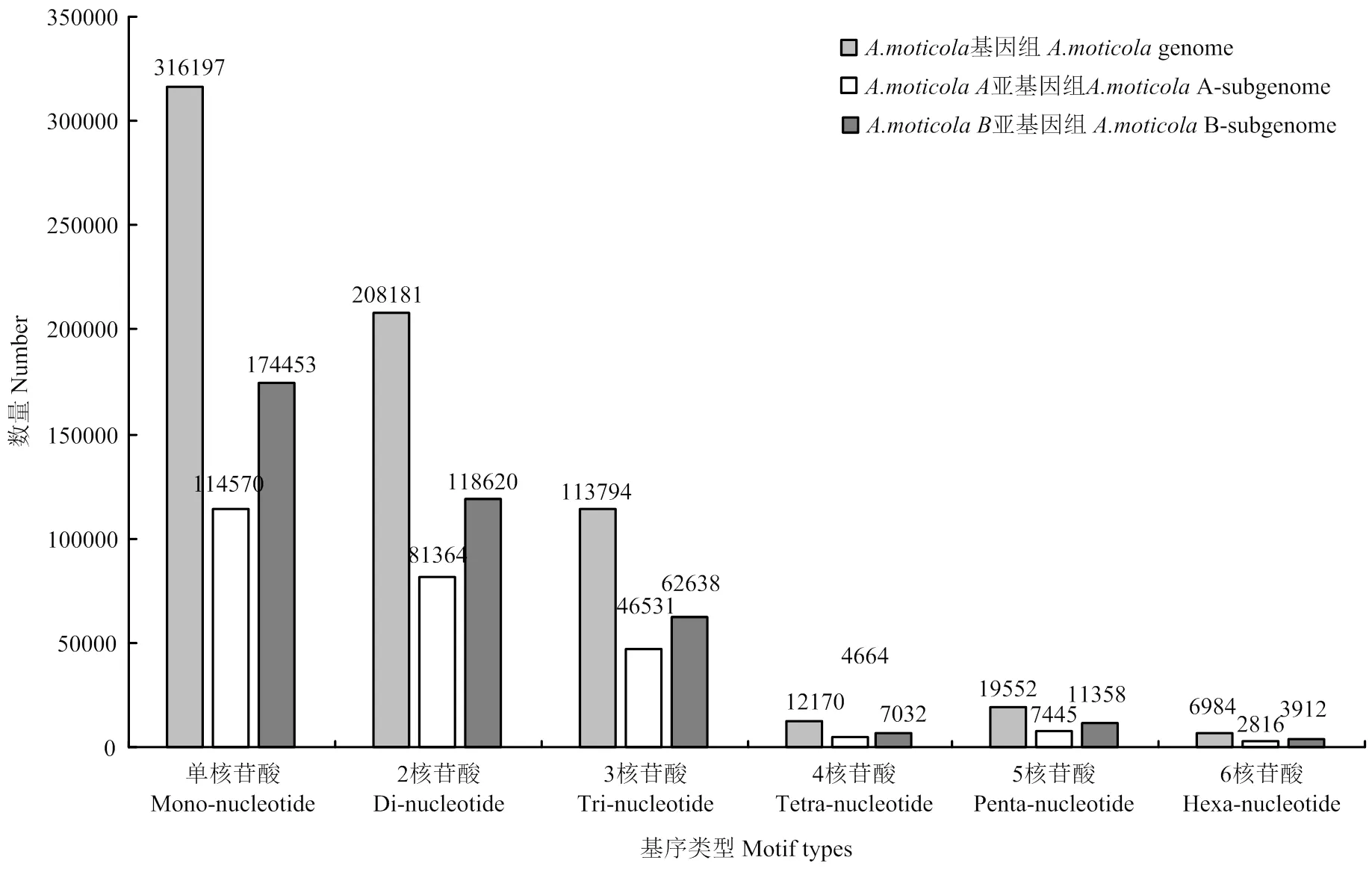
图1 A. monticola基因组SSR不同基序类型(1—6 bp)的数量分布

表3 SSR位点在A. monticola基因组上不同区域的数量分布
2.2 A.monticola全基因组SSR基序类型和结构
全基因组重复基元非常丰富,从基因组中鉴定出395个不同的重复基元(SSR motifs)。其中,在基因组中,单核苷酸有2种,二核苷酸有4种,三核苷酸有10种,四、五、六核苷酸分别有28、93和258种;的A基因组共鉴定出342种不同的重复基元,B基因组共搜索到356种不同的重复基元,其中,A、B亚基因组单核苷酸、二核苷酸、三核苷酸重复基元类型数目相同,分别为2、4和10种;四、五、六核苷酸分别存在特异的重复基元类型(表4)。四核苷酸重复基元类型:A基因组26种,特异性重复基元3种;B基因组25种,特异性重复基元2种。五核苷酸重复基元类型:A基因组81种,特异性重复基元6种;B基因组85种,特异性重复基元10种。六核苷酸重复基元:A基因组219种,特异性重复基元25种;B基因组230种,特异性重复基元36种。A/T是最丰富的重复基元,占基因组SSR的45.36%,其次是AT/AT、AAT/ATT、AG/CT、AAG/CTT和AC/GT,分别占基因组的19.26%、8.81%、8.71%、3.91%和2.41%。全基因组数量最多的20种SSR的重复基元如图2所示。
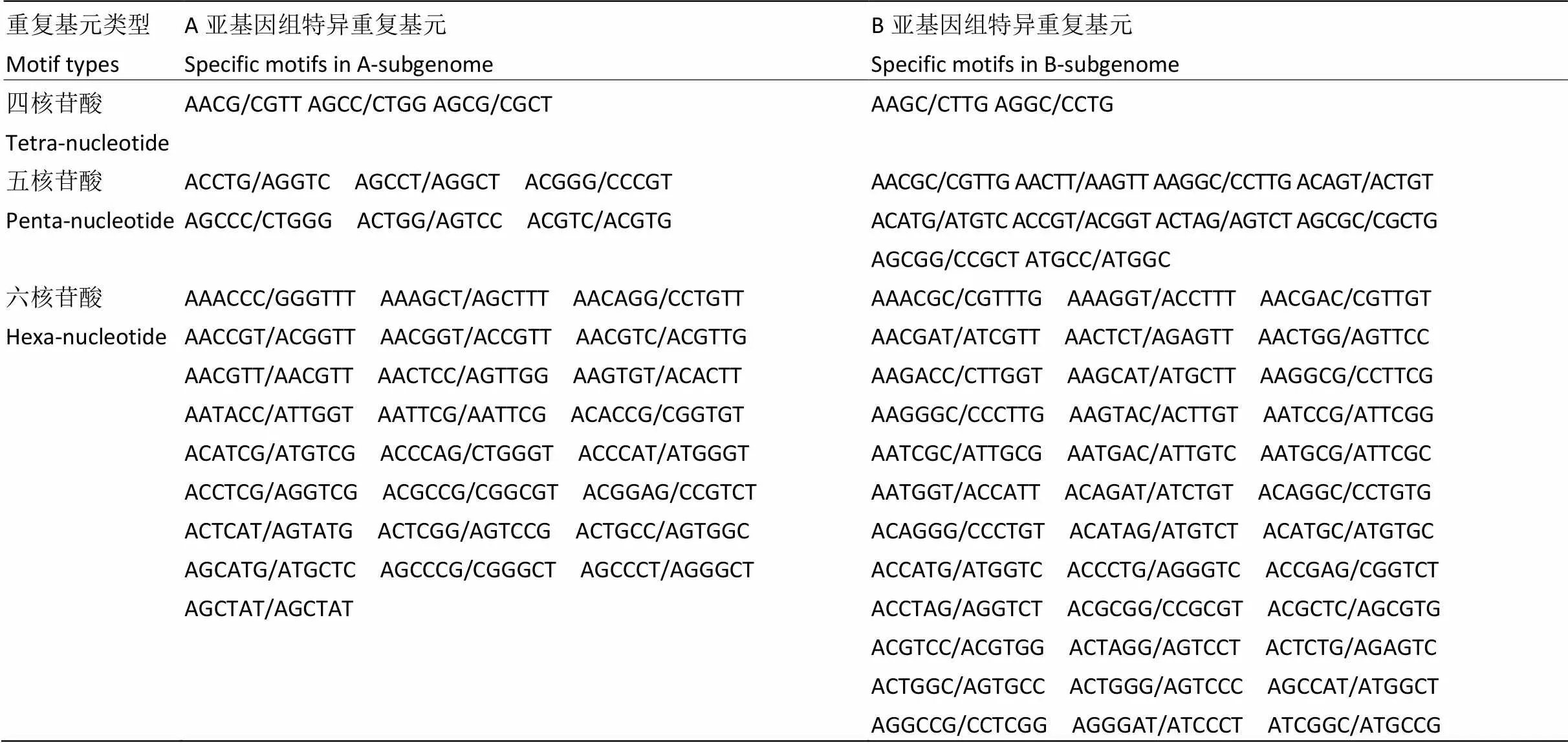
表4 A.monticola A、B亚基因组特异性重复基元类型
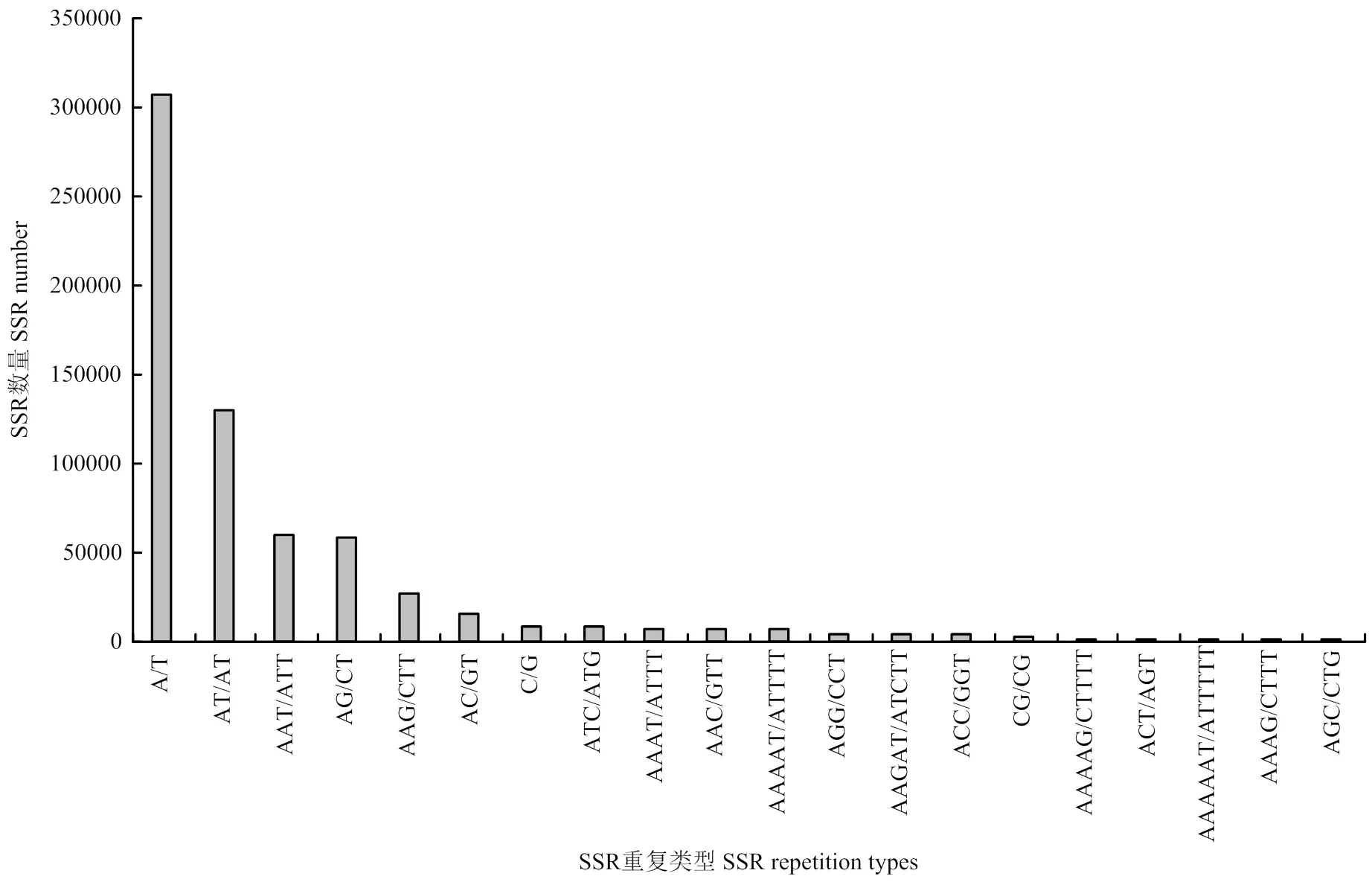
图2 A.monticola全基因组出现数量最多的20种SSR重复基元类型
2.3 A.monticola全基因组SSR碱基组成
在1—6个核苷酸的motifs中,单核苷酸重复基元数量最多的是A/T,二核苷酸最多的是AT/AT,三核苷酸最多的是AAT/ATT,四核苷酸最多的是AAAT/ATTT,其次是AAAAT/ATTTT和AAAAAT/ATTTTT,均为AT富集的重复基元,而GC富集的重复基元相对较少。
2.4 A.monticola全基因组SSR基序重复次数分布
从图3可以看出,重复基元的重复次数多集中在50次以内;不同类型的motif的重复次数差异很大;同一种类型motif的SSR位点,随着motif重复次数增加,SSR的数量逐渐降低,SSR重复次数越少,SSR数量越多。
2.5 A.monticola全基因组SSR在各染色体的分布
通过对各个染色体SSR数目及SSR频数进行统计(图4),各条染色体上均搜索到大量SSR。在20条染色体中,B03染色体上SSR数量最多,达到47 033个,平均307.18个SSR/Mb,平均每3.33 kb就会出现一个SSR;A08染色体中SSR密度最高,共15 257个SSR,平均403.19个SSR/Mb,平均2.54 kb就会出现一个SSR。其中A基因组A05染色体中SSR数量最多,其次是A01、A03、A10、A04、A09、A06、A02、A07和A08,A08染色体中SSR数量最少,但是密度最高,A03染色体SSR密度最低。B染色体组B03染色体SSR数量最多,其次是B09、B06、B04、B10、B02、B08、B01、B07和B05,且SSR密度最高,B05染色体中SSR数量最少,且SSR密度最低。B染色体组SSR数量远高于A染色体组。
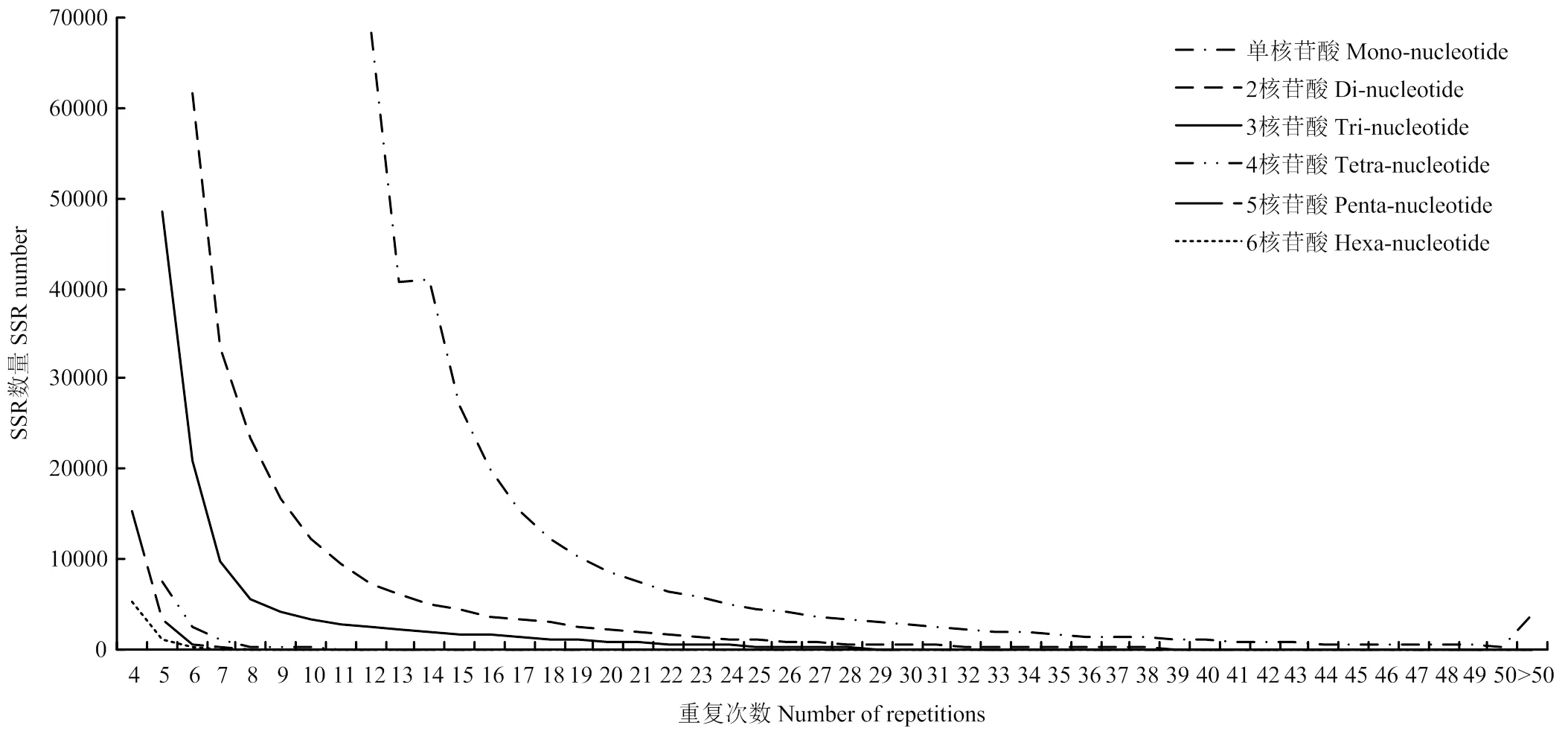
图3 不同类型的重复基元的重复次数分布
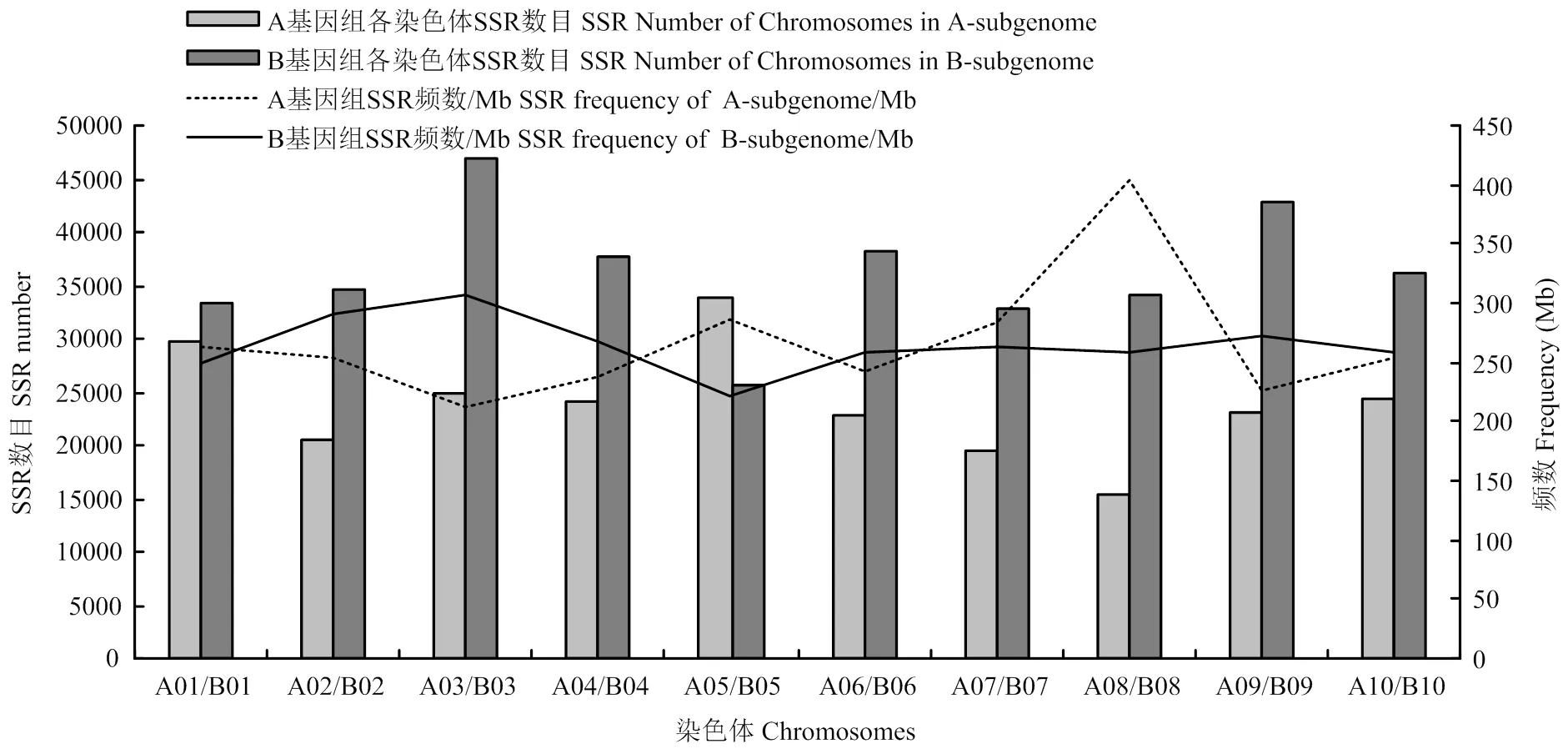
图4 A. monticola各条染色体SSR数目及SSR频数分布
2.6 A.monticola全基因组SSR引物开发,及单点SSR分析
利用SSR位点侧翼的保守序列,共设计出SSR引物192 303对,并利用这些标记进行电子定位,得到单位点标记96 828个(基因组中只有一个位点有扩增产物),检出率50.35%,单位点SSR在20条染色体上的分布密度呈现两端密集,中间稀疏的特点(图5和图6)。
图5 单位点SSR标记染色体上的数量分布
2.7 SSR引物的验证及通用性分析
在开发的单点SSR引物中,随机选取100对引物进行合成并实际扩增验证,在中能扩增出稳定清晰条带的有90对,用于验证的SSR引物在参试的4份花生材料中扩增条带表现出较好的通用性。如图7所示,目标条带在参试的4份花生材料中表现出不同的特点:引物A02-7和引物A04-20在4份材料中均能扩增出目标条带;引物A02-8和引物A03-12只能在和Tifrunner扩增出目的条带;引物A07-31、A07-32、A09-42、A09-43和A10-50能在和Tifrunner中扩增出目的条带,而在不能扩增出来;引物B07-82只能在、和Tifrunner中扩增出目的条带。引物A04-18只能在扩增出目的条带;引物B10-100只能在中扩增出目的条带。
3 讨论
3.1 A.monticola全基因组SSR分布特征
四倍体野生种花生基因组上SSR种类丰富,单核苷酸至六核苷酸均有分布,且数量上差异较大,全基因组SSR在基因组各区域的分布数量表明,大多数SSR分布在基因间区,然后是内含子区域,其次是外显子区域,最后是UTR区域,且5’UTR区域的SSR数量要高于3’UTR区域;在SSR重复碱基类型上,单碱基重复最为丰富,这与[45]、[45]、烟草[51]SSR位点以二核苷酸出现频率最高结果不一致,然后是二碱基重复,其次是三碱基重复,五碱基重复,四碱基重复,六碱基重复。SSR主要集中在一、二、三碱基重复,四、五、六碱基重复较少,这与拟南芥[49]、水稻[49]、玉米[55]、棉花[56]的结果相一致,表明进化水平相对较高。在SSR重复基序结构方面,单核苷酸A/T是最丰富的重复基元,占基因组SSR的45.36%,其次是AT/AT、AAT/ATT、AG/CT、AAG/CTT和AC/GT,分别占基因组的19.26%、8.81%、8.71%、3.91%和2.41%;二核苷酸最多的是AT/AT,此结果与[45]、[45]、烟草[51]基因组SSR研究结果相同,水稻[49]、玉米[55]、大豆[57]、小麦[58]中以AT/GC出现的频数最高有所不同;三核苷酸最多的是AAT/ATT,与拟南芥[50]重复基序AAG/TTC出现频数最高,水稻[49]、玉米[55]等CCG/GCC重复基序最丰富,ATT/TAA最少的研究结果不同。四核苷酸、五核苷酸、六核苷酸最多的依次是AAAT/ATTT、AAAAT/ATTTT、AAAAAT/ ATTTTT;对于SSR在各染色体上分布的数量和密度来看,在20条染色体中,B03染色体上SSR数量最多,达到47 033个,平均307.18个SSR/Mb,平均每3.33 kb就会出现一个SSR;A08染色体在20条染色体中最短,但SSR密度最高,共15 257个SSR,平均403.19个SSR/Mb,平均每2.54 kb就会出现一个SSR。
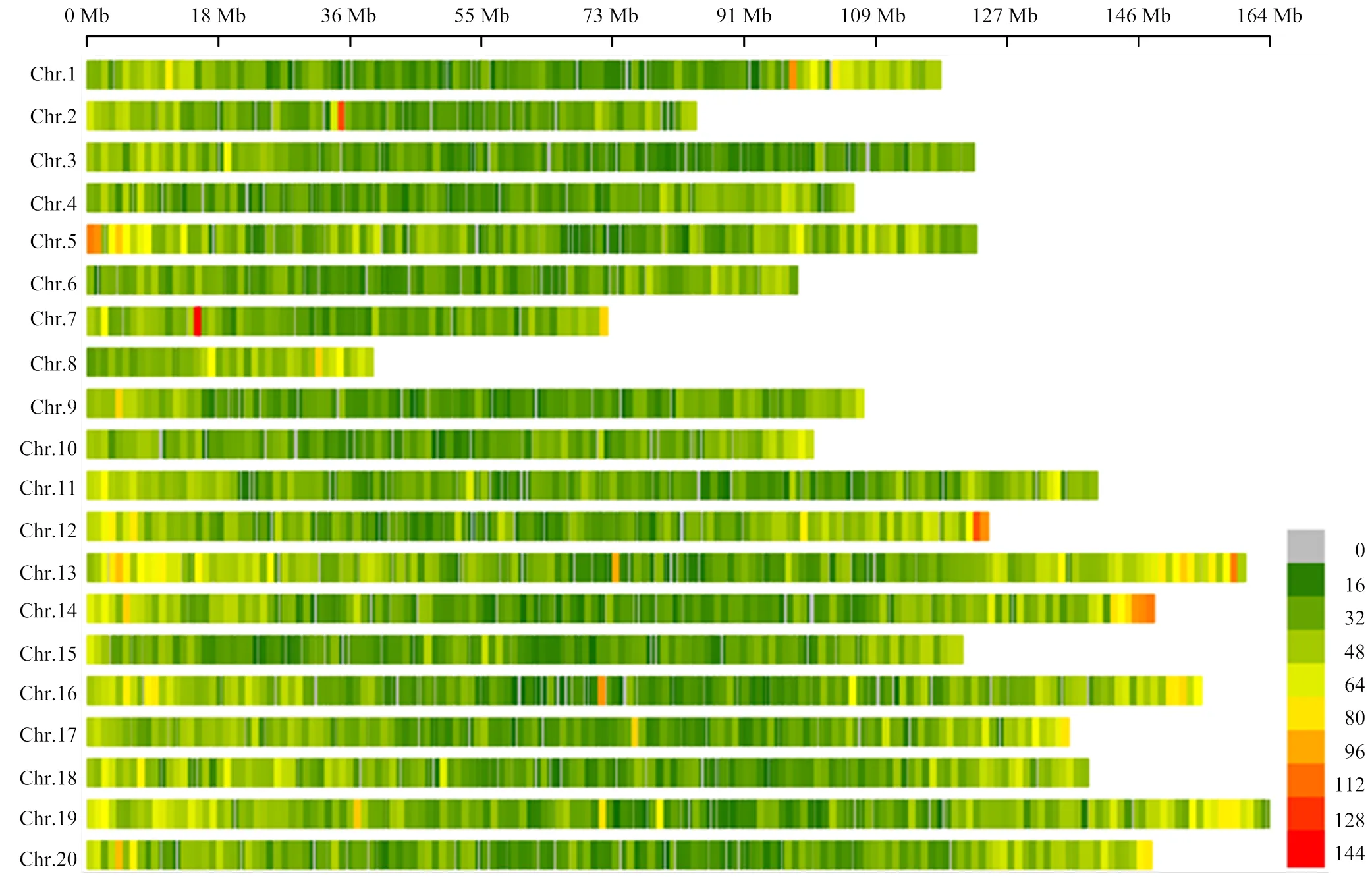
图6 SSR单位点标记在各染色体上的密度分布(窗口为1M)

M:DL 1000分子标记;1-4分别表示A. moticola、A. duranesis、A. ipaensis、Tifrunner;红色箭头表示目标条带
3.2 A. monticola与A. duranensis、A. ipaensis基因组SSR分布比较
本研究对四倍体野生种花生全基因组进行了SSR查找,共得到SSR位点676 878个。其中A基因组257 390个,密度为1个/4 kb,B基因组378 013个,密度为1个/3.9 kb,而密度为1个/8 kb,密度为1个/6.7 kb。总体水平上全基因组SSR数量比(A基因组)、(B基因组)SSR多,且密度更高。在重复基元数量分布方面,单碱基、二碱基、三碱基重复在A亚基因组和B亚基因组分别高于(A基因组)、(B基因组)。四碱基、五碱基、六碱基重复在A亚基因组数量高于,在B亚基因组数量低于;在重复基元分布比例方面,A、B亚基因组除单碱基比例分布高于、外二碱基、三碱基、四碱基、五碱基、六碱基比例均低于、;重复基元分布密度方面,在与基因组中SSR二核苷酸重复数量最多且密度最高,而基因组中单核苷酸重复数量最多且密度最高。出现这情况的原因有2种:(1)进化过程中SSR出现较大变异;(2)基因组序列组装存在问题。全基因组SSR均为AT富集的重复基元,而GC富集的重复基元相对较少,这与和基因组中SSR分布规律一致,这可能与打破GC键所需的能量要高于AT键所需的能量有关。基因组中较为丰富的重复序列的存在是导致花生基因组组装瓶颈的原因之一。
3.3 A.monticola基因组SSR标记的应用
前人研究表明,SSR引物在植物种属间具有极高的通用性[41,45,59]。花生全基因组序列的公布加速了基因组进化研究。本研究从基因组开发了SSR引物192 303对,其中96 828个单位点SSR标记,并在野生种花生、和栽培花生Tifrunner中初步验证了其通用性。所使用材料(AABB,2n=4x=40)是花生区组中唯一的异源四倍体野生种,被认为是栽培种花生亲缘关系最近的野生种[2];Tifrunner为栽培种;二倍体野生种和被认为是栽培花生的2个祖先,栽培花生A、B基因组的供体[1]。在漫长的进化过程中4份材料基因组序列会存在差异,材料的特殊性导致引物在4个类型基因组的品种中扩增条带存在较大差异。经比对,本研究开发的SSR引物与Zhao等[45]开发的引物有39 509对是重复序列,占比20.54%;其余152 794对为本研究新开发的SSR引物。由于在PCR过程中退火温度设置了不同的梯度,在扩增过程中会出现部分杂带(非特异性扩增)。本研究中SSR标记的开发对进一步研究花生属近缘种遗传多样性、基因组分子进化、染色体细胞学鉴定、品种真实性检测以及分子标记辅助选择育种等方面提供了重要的SSR分子标记库支撑。
4 结论
全基因组SSR位点各重复基元的数量、丰度、基序类型、重复次数以及碱基偏好性以及在各染色体上的分布均具有规律性。基因组上SSR种类丰富,单核苷酸至六核苷酸均有分布,且数量上差异较大,其中,单核苷酸重复基元数量最多,且最密,六核苷酸重复基元数量最少,出现频率最低,不同重复基元频数高低与核苷酸数量没有严格相关性。SSR多分布在基因间区,基因区内含子区域SSR数量最多;基因组中鉴定出395个不同的重复基元,A、B亚基因组具有其各自特异的重复基元类型;1—6个核苷酸的重复基元中,单个类型重复基元数量最多的均为AT富集的重复基元,而GC富集的重复基元相对较少;重复基元的重复次数多集中在50次以内,同一种类型重复基元的SSR位点,随着motif重复次数增加,SSR的数量逐渐降低;B亚基因组SSR数量远高于A亚基因组。全基因组SSR比、基因组SSR数量多,密度也更高,单核苷酸重复最丰富,与2个野生种二核苷酸数量最多的分布规律不同,单位点SSR在20条染色体上的分布密度呈现两端密集,中间稀疏的特点。基于全基因组SSR位点开发的引物,用于花生属的其他3个种表现出部分通用性,可用于花生近缘种的相关遗传分析。
[1] BERTIOLI D J, CANNON S B, FROENICKE L, HUANG G, FARMER A D, CANNON E K S, LIU X, GAO D, CLEVENGER J, DASH S, REN L, MORETZSOHN M C, SHIRASAWA K, HUANG W, VIDIGAL B, ABERNATHY B, CHU Y, NIEDERHUTH C E, UMALE P, ARAÚJO A C G, KOZIK A, KIM K D, BUROW M D, VARSHNEY R K, WANG X, ZHANG X, BARKLEY N, GUIMARÃES P M, ISOBE S, GUO B, LIAO B, STALKER H T, SCHMITZ R J, SCHEFFLER B E, LEAL-BERTIOLI S C M, XUN X, JACKSON S A, MICHELMORE R, OZIAS-AKINS P. The genome sequences ofand, the diploid ancestors of cultivated peanut., 2016, 48(4): 438-446.
[2] YIN D, JI C, MA X, LI H, ZHANG W, LI S, LIU F, ZHAO K, LI F, LI K, NING L, HE J, WANG Y, ZHAO F, XIE Y, ZHENG H, ZHANG X, ZHANG Y, ZHANG J. Genome of an allotetraploid wild peanut: a de novo assembly., 2018, 7(6): 1-9.
[3] WANG Z, WEBER J L, ZHONG G, TANKSLEY S D. Survey of plant short tandem DNA repeats., 1994, 88(1): 1-6.
[4] LEVINSON G, GUTMAN G A. Slipped-strand mispairing: a major mechanism for DNA sequence evolution., 1987, 4(3): 203-221.
[5] FIELD D, WILLS C. LONG, polymorphic microsatellites in simple organisms., 1996, 263(1367): 209-215.
[6] GUR-ARIE R, COHEN C J, EITAN Y, SHELEF L, HALLERMAN E M, KASHI Y. Simple sequence repeats in: abundance, distribution, composition, and polymorphism., 2000, 10(1): 62-71.
[7] TÓTH G, GÁSPÁRI Z, JURKA J. Microsatellites in different eukaryotic genomes: survey and analysis., 2000, 10(7): 967-981.
[8] LI Y C, KOROL A B, FAHIMA T, BEILES A, NEVO E. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review., 2002, 11(12): 2453-2465.
[9] KASHI Y, KING D G. Simple sequence repeats as advantageous mutators in evolution., 2006, 22(5): 253-259.
[10] 和小燕, 张建航, 刘婷, 王允, 马兴立, 张幸果, 殷冬梅. 花生F1代真伪杂种鉴定方法分析. 分子植物育种, 2018, 16(2): 477-483.
HE X Y, ZHANG J H, LIU T, WANG Y, MA X L, ZHANG X G, YIN D M. Analysis of identification method for hybrid F1generation of peanut., 2018, 16(2): 477-483. (in Chinese)
[11] 孙子淇, 张新友, 徐静, 张忠信, 刘华, 严玫, 董文召, 黄冰艳, 韩锁义, 汤丰收, 刘志勇. 河南省审定花生品种的指纹图谱构建. 作物学报, 2016, 42(10): 1448-1461.
SUN Z Q, ZHANG X Y, XU J, ZHANG Z X, LIU H, YAN M, DONG W Z, HUANG B Y, HAN S Y, TANG F S, LIU Z Y. DNA fingerprinting of peanut (L.) varieties released in henan province., 2016, 42(10): 1448-1461. (in Chinese)
[12] 胡晓辉, 毛瑞喜, 苗华荣, 石运庆, 崔凤高, 杨伟强, 陈静. 山东省46个花生品种SSR指纹图谱构建与遗传多样性分析. 核农学报, 2016, 30(10): 1925-1933.
HU X H, MAO R X, MIAO H R, SHI Y Q, CUI F G, YANG W Q, CHEN J. Construction of fingerprinting and analysis of genetic diversity with SSR markers for forty-six approved peanut cultivars from Shandong province., 2016, 30(10): 1925-1933. (in Chinese)
[13] 尹亮, 李双铃, 任艳, 石延茂, 袁美. 42个花生品种的SSR标记指纹图谱构建. 花生学报, 2017, 46(1): 8-13.
YIN L, LI S L, REN Y, SHI Y M, YUAN M. Construction of molecular fingerprint for 42 peanut varieties using SSR markers., 2017, 46(1): 8-13. (in Chinese)
[14] 韩柱强, 高国庆, 韦鹏霄, 唐荣华, 钟瑞春. 利用SSR标记分析栽培种花生多态性及亲缘关系. 花生学报, 2003(S1): 295-300.
HAN Z Q, GAO G Q, WEI P X, TANG R H, ZHONG R C. Analysis of DNA polymorphism and genetic relationships in cultivated peanut (L.) using microsatellite markers., 2003(S1): 295-300. (in Chinese)
[15] 任小平, 张晓杰, 廖伯寿, 雷永, 黄家权, 陈玉宁, 姜慧芳. ICRISAT花生微核心种质资源SSR标记遗传多样性分析. 中国农业科学, 2010, 43(14): 2848-2858.
REN X P, ZHANG X J, LIAO B S, LEI Y, HUANG J Q, CHEN Y N, JIANG H F. Analysis of genetic diversity in ICRISAT mini core collection of peanut (L.) by SSR markers., 2010, 43(14): 2848-2858. (in Chinese)
[16] 詹世雄, 郑奕雄, 刘冠明, 张平湖, 杨灵, 庄东红. 基于SSR标记的花生品种遗传多样性分析. 中国油料作物学报, 2014, 36(2): 269-274.
ZHAN S X, ZHENG Y X, LIU G M, ZHANG P H, YANG L, ZHUANG D H. Genetic diversity in peanut cultivars based on SSR markers., 2014, 36(2): 269-274. (in Chinese)
[17] 王燕龙, 单雷, 付春, 徐平丽, 姜言生, 柳展基, 曲志才, 唐桂英. 不同SSR标记检测技术及其在花生栽培种遗传多样性分析中的应用. 植物遗传资源学报, 2014, 15(1): 96-105.
WANG Y L, SHAN L, FU C, XU P L, JIANG Y S, LIU Z J, QU Z C, TANG G Y. Different SSR detection techniques and their application in genetic diversity analysis of peanut (L.) cultivars., 2014, 15(1): 96-105. (in Chinese)
[18] REN X, JIANG H, YAN Z, CHEN Y, ZHOU X, HUANG L, LEI Y, HUANG J, YAN L, QI Y, WEI W, LIAO B. Genetic diversity and population structure of the major peanut (L.) cultivars grown in China by SSR markers., 2014, 9(2): e88091.
[19] VARSHNEY R K, BERTIOLI D J, MORETZSOHN M C, VADEZ V, KRISHNAMURTHY L, ARUNA R, NIGAM S N, MOSS B J, SEETHA K, RAVI K, HE G, KNAPP S J, HOISINGTON D A. The first SSR-based genetic linkage map for cultivated groundnut (L.)., 2009, 118(4): 729-739.
[20] 刘华. 栽培花生产量和品质相关性状遗传分析与QTL定位研究[D]. 郑州: 河南农业大学, 2011.
LIU H. Inheritance of main traits related to yield and quality, and their QTL mapping in peanut (L.)[D]. Zhengzhou: Henan Agricultural University, 2011. (in Chinese)
[21] SHIRASAWA K, BERTIOLI D J, VARSHNEY R K, MORETZSOHN M C, LEAL-BERTIOLI S C M, THUDI M, PANDEY M K, RAMI J F, FONCEKA D, GOWDA M V C, QIN H , GUO B, HONG Y, LIANG X, HIRAKAWA H , TABATA S, ISOBE S. Integrated consensus map of cultivated peanut and wild relatives reveals structures of the A and B genomes ofand divergence of the legume genomes., 2013, 20(2): 173.
[22] PANDEY M K, WANG M L, QIAO L, FENG S, KHERA P, WANG H, TONNIS B, BARKLEY N A, WANG J, HOLBROOK C C, CULBREATH A K, VARSHNEY R K, GUO B. Identification of QTLs associated with oil content and mapping FAD2 genes and their relative contribution to oil quality in peanut (L.)., 2014, 15(1): 1-14.
[23] LI Y, LI L, ZHANG X, ZHANG K, MA D, LIU J, WANG X, LIU F, WAN Y. QTL mapping and marker analysis of main stem height and the first lateral branch length in peanut (L.)., 2017, 213(2): 57.
[24] HOPKINS M S, CASA A M, WANG T, MITCHELL S E, DEAN R E, KOCHERT G D, KRESOVICH S. Discovery and characterization of polymorphic simple sequence repeats (SSRs) in peanut., 1999, 39(4): 1243-1247.
[25] HE G, MENG R, NEWMAN M, GAO G, PITTMAN R N, PRAKASH C S. Microsatellites as DNA markers in cultivated peanut (L.)., 2003, 3(1): 3.
[26] FERGUSON M E, BUROW M D, SCHULZE S R, BRAMEL P J, PATERSON A H, KRESOVICH S, MITCHELL S. Microsatellite identification and characterization in peanut (L.)., 2004, 108(6): 1064-1070.
[27] MORETZSOHN M D C, HOPKINS M S, MITCHELL S E, KRESOVICH S, VALLS J F M, FERREIRA M E. Genetic diversity of peanut (L.) and its wild relatives based on the analysis of hypervariable regions of the genome., 2004, 4(1): 11.
[28] MORETZSOHN M C, LEOI L, PROITE K, GUIMARÃES P M, LEAL-BERTIOLI S C M, GIMENES M A, MARTINS W S, VALLS J F M, GRATTAPAGLIA D, BERTIOLI D J. A microsatellite-based, gene-rich linkage map for the AA genome of(Fabaceae)., 2005, 111(6): 1060-1071.
[29] CUC L M, MACE E S, CROUCH J H, QUANG V D, LONG T D, VARSHNEY R K. Isolation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut ()., 2008, 8(1): 55.
[30] YUAN M, GONG L, MENG R, LI S, DANG P, GUO B, HE G. Development of trinucleotide (GGC)n SSR markers in peanut (L.)., 2010, 13(6): 5-6.
[31] SHIRASAWA K, KOILKONDA P, AOKI K, HIRAKAWA H, TABATA S, WATANABE M, HASEGAWA M, KIYOSHIMA H, SUZUKI S, KUWATA C, NAITO Y, KUBOYAMA T, NAKAYA A, SASAMOTO S, WATANABE A, KATO M, KAWASHIMA K, KISHIDA Y, KOHARA M, KURABAYASHI A, TAKAHASHI C, TSURUOKA H, WADA T, ISOBE S. In silico polymorphism analysis for the development of simple sequence repeat and transposon markers and construction of linkage map in cultivated peanut., 2012, 12(1): 80.
[32] MACEDO S E, MORETZSOHN M C, M LEAL-BERTIOLI S C, ALVES D M, GOUVEA E G, AZEVEDO V C, BERTIOLI D J. Development and characterization of highly polymorphic long TC repeat microsatellite markers for genetic analysis of peanut., 2012, 5(1): 86.
[33] LUO M, DANG P, GUO B Z, HE G, HOLBROOK C C, BAUSHER M G, LEE R D. Generation of expressed sequence tags (ESTs) for gene discovery and marker development in cultivated peanut., 2005, 45(1): 346-353.
[34] PROITE K, LEAL-BERTIOLI S C , BERTIOLI D J, MORETZSOHN M C, SILVA F R D, MARTINS N F, GUIMARÃES P M. ESTs from a wildspecies for gene discovery and marker development., 2007, 7(1): 7.
[35] LIANG X, CHEN X, HONG Y, LIU H, ZHOU G, LI S, GUO B. Utility of EST-derived SSR in cultivated peanut (L.) andwild species., 2009, 9(1): 35.
[36] WANG J, PAN L, YANG Q, YU S. Development and characterization of EST-SSR markers from NCBI and cDNA library in cultivated peanut (L.)., 2010, 1(6):30-33.
[37] KOILKONDA P, SATO S, TABATA S, SHIRASAWA K, HIRAKAWA H, SAKAI H, SASAMOTO S, WATANABE A, WADA T, KISHIDA Y, TSURUOKA H, FUJISHIRO T, YAMADA M, KOHARA M, SUZUKI S, HASEGAWA M, KIYOSHIMA H, ISOBE S. Large-scale development of expressed sequence tag-derived simple sequence repeat markers and diversity analysis inspp., 2012, 30(1): 125-138.
[38] GUO Y, KHANAL S, TANG S, BOWERS J E, HEESACKER A F, KHALILIAN N, NAGY E D, ZHANG D, TAYLOR C A, STALKER H T, OZIAS-AKINS P, KNAPP S J. Comparative mapping in intraspecific populations uncovers a high degree of macrosynteny between A-and B-genome diploid species of peanut., 2012, 13(1): 608.
[39] BOSAMIA T C, MISHRA G P, THANKAPPAN R, DOBARIA J R. Novel and stress relevant EST derived SSR markers developed and validated in peanut., 2015, 10(6): e0129127.
[40] HUANG L, WU B, ZHAO J, LI H, CHEN W, ZHENG Y, REN X, CHEN Y, ZHOU X, LEI Y, LIAO B, JIANG H. Characterization and transferable utility of microsatellite markers in the wild and cultivated Arachis species., 2016, 11(5): e0156633.
[41] HE G, WOULLARD F E, MARONG I, GUO B Z. Transferability of soybean SSR markers in peanut (L.)., 2006, 33(1): 22-28.
[42] WANG H, PENMETSA R V, YUAN M, GONG L, ZHAO Y, GUO B, FARMER A D, ROSEN B D, GAO J, ISOBE S, BERTIOLI D J, VARSHNEY R K, COOK D R, HE G. Development and characterization of BAC-end sequence derived SSRs, and their incorporation into a new higher density genetic map for cultivated peanut (L.)., 2012, 12(1): 10.
[43] ZHANG J, LIANG S, DUAN J, WANG J, CHEN S, CHENG Z, ZHANG Q, LIANG X, LI Y. De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in peanut (L.)., 2012, 13(1): 90.
[44] PENG Z, GALLO M, TILLMAN BL, ROWLAND D, WANG J. Molecular marker development from transcript sequences and germplasm evaluation for cultivated peanut (L.)., 2016, 291(1): 363-381.
[45] ZHAO C, QIU J, AGARWAL G, WANG J, REN X, XIA H, GUO B, MA C, WAN S, BERTIOLI D J, VARSHNEY R K, PANDEY M K, WANG X. Genome-wide discovery of microsatellite markers from diploid progenitor species,and, and their application in cultivated peanut ()., 2017, 8: 1209.
[46] LUO H, XU Z, LI Z, LI X, LV J, REN X, HUANG L, ZHOU X, CHEN Y, YU J, CHEN W, LEI Y, LIAO B, JIANG H. Development of SSR markers and identification of major quantitative trait loci controlling shelling percentage in cultivated peanut (L.)., 2017, 130(8): 1635-1648.
[47] ZHAO Y, PRAKASH C S, HE G. Characterization and compilation of polymorphic simple sequence repeat (SSR) markers of peanut from public database., 2012, 5(1): 362.
[48] SHIRASAWA K, ISOBE S, TABATA S, HIRAKAWA H. Kazusa Marker DataBase: a database for genomics, genetics, and molecular breeding in plants., 2014, 64(3): 264-271
[49] LAWSON M J, ZHANG L Q. Distinct patterns of SSR distribution in theand rice genomes., 2006, 7(2): R14.1-R14.11.
[50] CAVAGNARO P F, SENALIK D A, YANG L, SIMON P W, HARKINS T T, KODIRA C D, HUANG S, WENG Y. Genome-wide characterization of simple sequence repeats in cucumber (L.)., 2010, 11(1): 569.
[51] 童治军, 焦芳婵, 肖炳光. 普通烟草及其祖先种基因组 SSR 位点分析. 中国农业科学, 2015, 48(11): 2108-2117.
TONG Z J, JIAO F C, XIAO B G. Analysis of SSR loci ingenome and its two ancestral species genome., 2015, 48(11): 2108-2117. (in Chinese)
[52] SONAH H, DESHMUKH R K, SHARMA A, SINGH V P, GUPTA D K, GACCHE R N, RANA J C, SINGH N K, SHARMA T R. Genome-wide distribution and organization of microsatellites in plants: an insight into marker development in., 2011, 6(6): e2129
[53] 蔡斌, 李成慧, 姚泉洪, 周军, 陶建敏, 章镇. 葡萄全基因组SSR分析和数据库构建. 南京农业大学学报, 2009, 32(4): 28-32.
CAI B, LI C H, YAO Q H, ZHOU J, TAO J M, ZHANG Z. Analysis of SSRs in grape genome and development of SSR database., 32(4): 28-32. (in Chinese)
[54] YU J, DOSSA K, WANG L, ZHANG Y, WEI X, LIAO B, ZHANG X. PMDBase: a database for studying microsatellite DNA and marker development in plants., 2017, 45(Database issue): D1046-D1053.
[55] 原志敏. 玉米全基因组 SSRs 分子标记开发与特征分析[D]. 雅安: 四川农业大学, 2013.
YUAN Z M. Development and characterization of SSR markers providing genome-wide coverage and high resolution in maize[D]. Yaan: Sichuan Agricultural University, 2013. (in Chinese)
[56] KANTETY R V, ROTA M L, MATTHEWS D E, SORRELLS M E. Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat., 2002, 48: 501-510.
[57] SONG Q, JIA G, ZHU Y, GRANT D, NELSON R T, WANG E Y, HYTEN D L, CREGAN P B. Abundance of SSR motifs and development of candidate polymorphic SSR markers (BARCSOYSSR1.0) in soybean., 2010, 50(5): 1950-1960.
[58] 郑燕, 张耿, 吴为人. 禾本科植物微卫星序列的特征分析和比较. 基因组学与应用生物学, 2011, 30: 513-520.
ZHENG Y, ZHANG G, WU W R. Characterization and comparison of microsatellites in gramineae., 2011, 30: 513-520. (in Chinese)
[59] 陈明丽, 王兰芬, 武晶, 张晓艳, 杨广东, 王述民. 普通菜豆基因组SSR标记开发及在豇豆和小豆中的通用性分析. 作物学报, 2014, 40(5): 924-933.
CHEN M L, WANG L F, WU J, ZHANG X Y, YANG G D, WANG S M. Development of genomic SSR markers in common bean and their transferability in cowpea and adzuki bean., 2014, 40(5): 924-933. (in Chinese)
Development and Characterization of Whole Genome SSR in Tetraploid Wild Peanut ()
Wang YuLong1,2, Huang BingYan2, Wang SiYu2,3, Du Pei2, Qi FeiYan2, Fang YuanJin2, Sun ZiQi2, Zheng Zheng2, Dong WenZhao2, Zhang XinYou1,2
(1College of Agriculture, Henan University of Science and Technology, Luoyang 471023, Henan;2Industrial Crops Research Institute, Henan Academy of Agricultural Sciences/Key Laboratory of Oil Crops in Huanghuaihai Plain/Henan Provincial Key Laboratory for Oil Crops Improvement, Zhengzhou 450002;3School of Life Sciences, Zhengzhou University, Zhengzhou 450001)
【】We aimed to identify simple sequence repeat (SSR) loci throughout the genome of tetraploid wild peanut(AABB, 2n = 4x = 40), to identify their distribution characteristics, and to develop and validate SSR primers. These markers have potential uses in genetic evolution analyses and in the development of molecular markers for important traits in peanut.【】Using the bioinformatics software MISA, we searched for SSR loci in the whole genome sequence of, which was downloaded from the GigaScience database of the BGI. One hundred SSR loci were randomly selected and primers were designed and synthesized. The primers were used to amplify products from four differentgenomes, and the products were analyzed by polyacrylamide gel electrophoresis (PAGE).【】A total of 676 878 SSRs were found in the genome of tetraploid wild peanutin 5 127 scaffolds (average, one SSR per 3.8 kb). The SSRs ranged from single nucleotides to hexanucleotides. Single nucleotide SSRs were significantly more abundant than hexanucleotide SSRs. Single, double, and triple nucleotide SSRs were predominant, accounting for 94.28% of all the SSRs. Single nucleotide SSRs accounted for the largest proportion of total SSRs (46.71%) and showed the highest density. Hexanucleotide SSRs accounted for the smallest proportion and showed the sparsest density. Most SSRs were located in intergenic regions, and most of the SSRs in gene sequences were located in introns. A total of 395 different repeat motifs were identified in the whole genome, of which 342 were in the A-subgenome and 356 were in the B-subgenome. The most abundant repeat motif was A/T. The most abundant repeat motifs for SSRs with 1–6 nucleotides were A/T, AT/AT, AAT/ATT, AAAT/ATTT, AAAAT/ATTTT, and AAAAAT/ATTTTT, respectively. There were less than 50 of each type of SSR repeat motif, but the number of each type of SSR motif varied greatly. The number of each type of SSRs repeat motif decreased with increasing number of nucleotides in the motif. Chromosome B03 had the most SSRs, and chromosome A08 showed the highest density of SSRs. We designed 192 303 pairs of SSR primers, and the detection rate of single-locus SSR markers was 50.35%. The distribution of SSR markers in the genome was dense at both ends and sparse in the middle. Among the 100 synthesized primer pairs, 90 pairs amplified stable and clear bands fromgenomic DNA. The bands amplified from four different peanut genomic DNAs showed different characteristics. 【】Thegenome was rich in SSRs ranging from single nucleotides to hexanucleotides. Single nucleotide repeats were the most abundant and densely distributed, and hexanucleotides showed the lowest frequency and the sparsest distribution. There was no strict correlation between the frequency of different repeats and the repeat type. The A-subgenome and B-subgenome had their own specific SSRs. The AT-enriched repeat motifs were the most abundant, while GC-enriched repeat motifs showed much lower frequencies. The number of SSRs with the same type of repeat motif decreased with increasing numbers of nucleotides in the motif. Compared with the genomes of the two diploid wild species, the tetraploid genome ofhad more SSRs, a higher density of SSRs, and a different SSR distribution pattern. Preliminary validation analyses showed that the SSR primers designed in this study shared certain universal properties among fourgenomes.
peanut;; whole genome sequence; SSR locus; motif
2019-03-17;
2019-05-15
国家花生产业技术体系(CARS-13)、河南省重大科技专项(16110011100)、河南省花生产业技术体系(2012-5)
王玉龙,E-mail:wangyulong0724@126.com。
张新友,E-mail:haasxinyou@163.com
(责任编辑 李莉)
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
兵工学报(2022年2期)2022-05-22
兵工学报(2021年4期)2021-06-19
教学考试(高考生物)(2020年6期)2020-11-23
兵工学报(2020年12期)2020-02-06
食品与生物技术学报(2020年8期)2020-01-06
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
