
神经节苷脂氟化寡糖在大肠杆菌中的生物合成
2019-08-08 03:32刘新平谭玉萌张雪冯雁杨广宇
生物技术通报 2019年8期
刘新平 谭玉萌 张雪 冯雁 杨广宇
(上海交通大学生命科学技术学院,上海 200240)
神经节苷脂是由含唾液酸的寡糖和神经酰胺组成的鞘糖脂类化合物,广泛存在于所有脊椎动物细胞中,尤其是在神经细胞中含量丰富。它们在细胞识别、细胞免疫、细胞凋亡等多种细胞生理活动中发挥重要作用[1-2]。GM1是目前研究最深入的神经节苷脂,在神经退行性疾病如外伤性中枢神经系统损伤、早期阿尔兹海默症等疾病中体现了重要的药物活性[3]。GM2和GM3广泛参与细胞间信号转导、细胞增殖、细胞黏附和细胞运动等过程。在多种肿瘤细胞中,GM2和GM3表达增强并参与肿瘤的迁移过程,成为药物开发的靶标[4-5]。因此,发展神经节苷脂的高效制备技术,对于研究此类化合物的生理功能、作用机制及促进新药开发均具有重要意义。
神经节苷脂结构复杂,难以利用传统化学催化方法进行合成。目前其主要生产来源是从天然生物组织如猪脑、牛脑中进行提取,但仍存在成本高、收率低、提取过程污染严重等局限[6-7]。近年来,利用生物系统进行神经节苷脂的人工合成研究取得很大进展。例如Yu等[8-9]开发了以糖基转移酶为基础的一锅多酶(One-pot multienzyme,OPME)催化体系,但该体系中需要近10种酶协同作用,酶的表达、提取步骤繁多且需要消耗大量昂贵的糖核苷酸等底物,难以实现大量合成。Fierfort和Antoine等[10-11]利用代谢工程手段,在大肠杆菌体内构建了一系列用于神经节苷脂寡糖模块合成的细胞工厂,但是由于缺少寡糖链和神经酰胺链的连接方法,无法实现完整神经节苷脂的合成。本实验在前期研究工作中,基于合成生物学理念提出了新的神经节苷脂合成体系[12](图1)。该体系主要分为3步:(1)以氟化乳糖为底物,利用糖基转移酶催化合成神经节苷脂氟化寡糖;(2)利用糖苷合成酶EGCase独特的催化活性,催化神经节苷脂氟化寡糖与鞘氨醇组装形成糖基鞘氨醇;(3)在鞘糖脂N-酰化酶SCDase催化下,糖基鞘氨醇与硬脂酸缩合形成完整的神经节苷脂。
神经节苷脂氟化寡糖的高效合成是上述整个合成体系的关键步骤。本研究以Escherichia coliJM107为出发菌株,该菌株敲除了半乳糖苷酶基因lacZ和唾液酸醛缩酶基因nanA,避免了氟化乳糖和唾液酸底物在胞内被大量降解。在此基础上,在大肠杆菌中引入脑膜炎奈瑟氏菌(Neisseria meningitidis)来源的CMP-NeuAc合成酶(neuA)[13]、空肠弯曲杆菌(Campylobacter jejuni)来源的 α 2,3-NeuAc转移酶(cst)[14]、β 1,4-GalNAc转移(cgtA)[15]、β 1,3-Gal转移酶(cgtB)[16]和 UDP-GlcNAc 异构酶(gne)[17],从而构建以氟化乳糖和唾液酸为底物的神经节苷脂GM3、GM2和GM1氟化寡糖生物合成途径(图2)。
1 材料与方法
1.1 材料
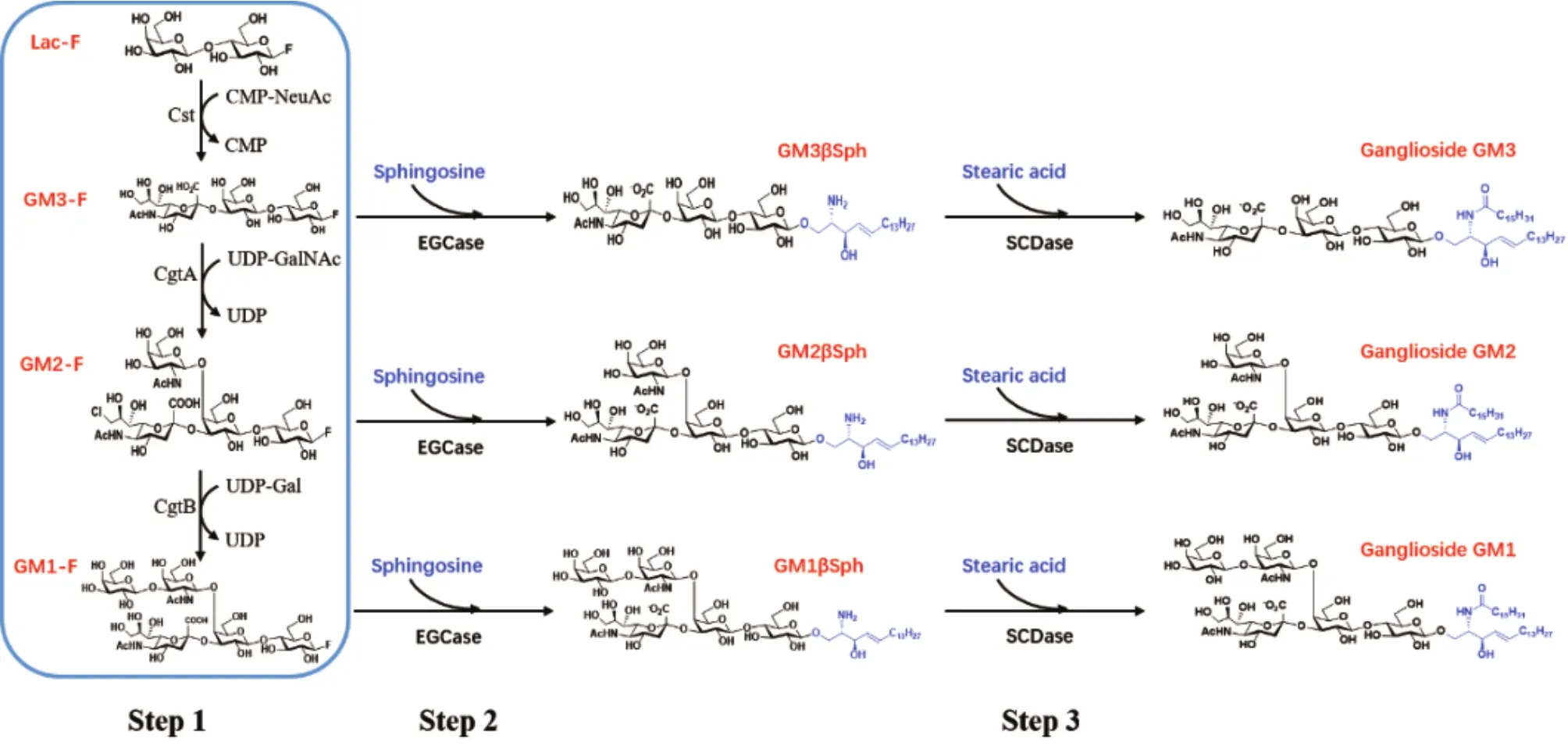
图1 完整神经节苷脂GM3、GM2和GM1合成体系
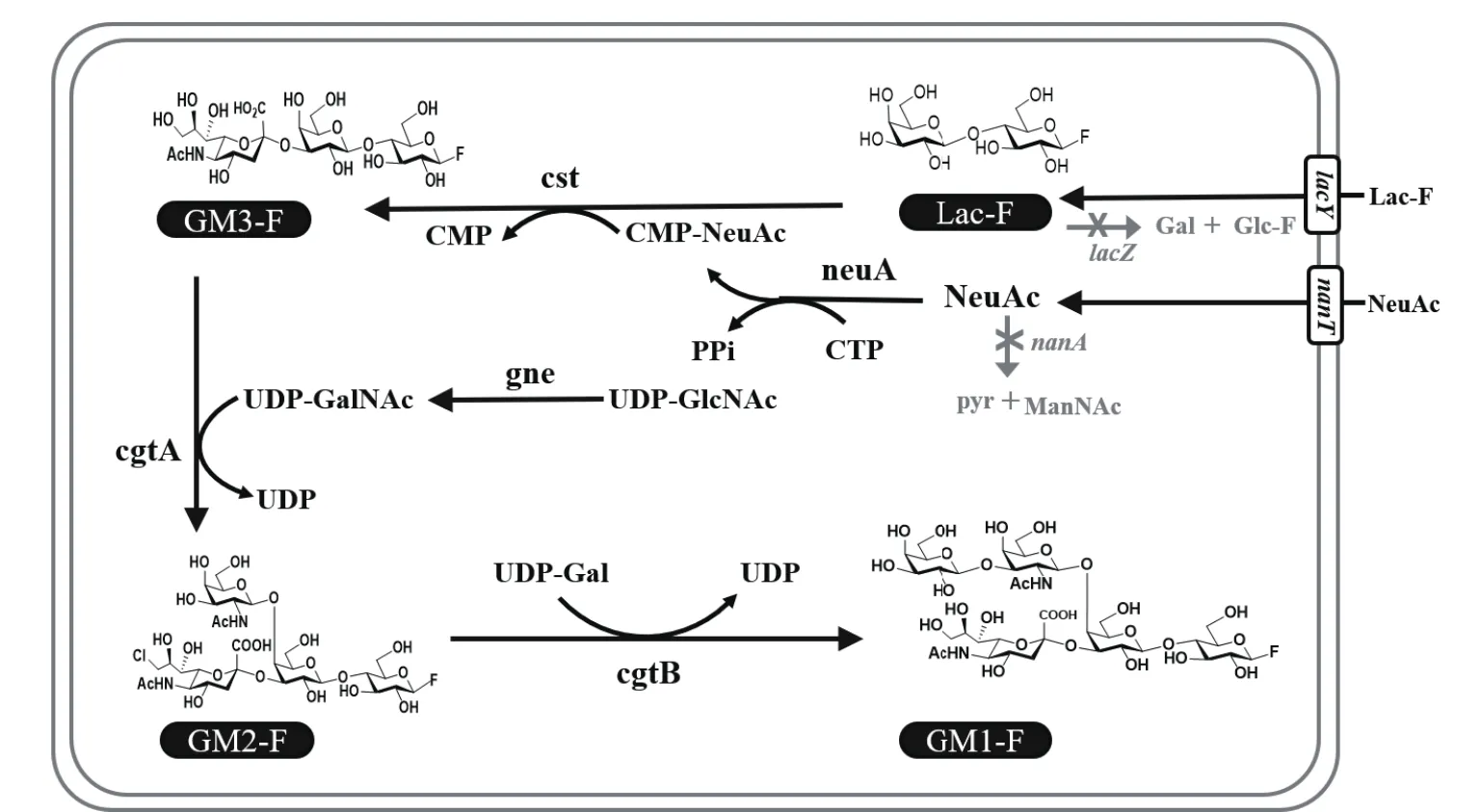
图2 神经节苷脂GM3、GM2、GM1氟化寡糖生物合成途径
1.1.1 菌种与质粒 敲除了nanA和lacZ两个基因的Escherichia coliJM107作为出发菌株用于人工生物途径的构建,该菌株由本实验室保存。Escherichia coliDH5α用于载体的构建,购自全式金生物有限公司。质粒pUC18由本实验室保存,质粒pBBR1MSC3,pACYC184购自湖南丰晖生物技术有限公司。
1.1.2 试剂及培养基 PrimeSTAR Max Premix DNA聚合酶购于日本Takara公司;对氨基苯甲酸、氰基硼氢化钠、醋酸钠等衍生试剂购自阿拉丁试剂有限公司;相关氟化寡糖由本实验室保存;唾液酸、甲酸、甲酸铵等购自Sigma公司;乙腈等液相用品购自上海安谱实验科技股份有限公司。LB培养基氯化钠10 g/L,胰蛋白胨10 g/L,酵母浸粉5 g/L。
1.2 方法
1.2.1 重组质粒的构建 目的基因neuA(GenBank:U60146.1),cst(GenBank:AF130466.2),gne(GenBank:905422),cgtA(GenBank:AF401528.1),cgtB(GenBank:AF130984.1)均由本实验室保存。重组载体采用重叠延伸PCR(Prolonged Overlap Extension cloning)的方法构建而成[18]。以重组质粒pUC18-neuA-cst的构建为例说明POEPCR的具体方法。在引物设计时,基因片段与载体之间,串联基因之间均保留25 bp左右的同源臂,分别使用特异性引物IF1-neuA/IR1-neuA扩增第一个插入片段neuA基因,引物IF2-cst/IR2-cst扩增第二个插入片段cst基因,引物VF1-pUC18/ VR1-pUC18进行反向PCR扩增来制备线性化载体。扩增获得的基因片段和线性载体进行胶回收后,以基因片段和线性化载体互为模板和引物进行下一轮的PCR扩增。PCR 产物直接转化Escherichia coliDH5α感受态细胞,在含氨苄抗性的平板上挑取单克隆进行培养,培养的菌液提取质粒并测序验证,获得重组质粒pUC18-neuA-cst。另两个重组质粒的构建方法同上,gne和cgtA基因串联克隆至pBBR1MSC3载体,获得重组质粒pBBR1MCS3-gne-cgtA,cgtB基因克隆至pACYC184载体,获得重组质粒pACYC184-cgtB。引物设计见表1。
1.2.2 工程菌株的构建 将单质粒pUC18-neuA-cst转入Escherichia coliJM107感受态细胞中,利用氨苄抗性平板筛选得到GM3氟化寡糖合成的菌株Strain-GM3。将双质粒pUC18-neuA-cst和pBBR1MCS3-gnecgtA电转化至Escherichia coliJM107感受态细胞中,利用氨苄和四环素双抗平板筛选得到GM2氟化寡糖合成的菌株Strain-GM2。将3质粒pUC18-neuA-cst、pBBR1MCS3-gne-cgtA和pACYC184-cgtB共同转化Escherichia coliJM107菌株,利用氨苄、四环素和氯霉素三抗平板筛选获得GM1氟化寡糖合成的菌株Strain-GM1。
1.2.3 工程菌株的发酵 挑取Strain-GM3、Strain-GM2、Strain-GM1的单克隆分别接种于相对应抗性的4 mL液体LB培养基中,于37℃恒温摇床中震荡培养过夜。按1%的接菌量将3种新鲜菌液接种至200 mL含相应抗性的LB液体培养基中,于37℃的恒温摇床中震荡培养至OD600为0.8左右。加入终浓度为50 mg/L的IPTG诱导剂、终浓度为2 g/L的氟化乳糖和终浓度为1.5 g/L的唾液酸,于30℃恒温摇床中震荡培养90 h,并在培养过程中取样分析。

表1 重组载体构建引物设计
1.2.4 发酵产物的检测及鉴定 使用HPLC柱前衍生的方法对GM3、GM2、GM1氟化寡糖进行定量检测。利用对氨基苯甲酸衍生化试剂对3种寡糖还原端的醛基进行衍生化,衍生试剂为:10 mg/mL对氨基苯甲酸和10 mg/mL氰基硼氢化钠溶于4%(W/V)醋酸钠-甲醇缓冲液中。取不同发酵时间下的新鲜发酵液,超声破碎收集上清液,加入4倍体积的衍生试剂,置于70℃的恒温水浴中保温45 min后终止反应,12 000 r/min离心15 min,吸取上清液用于HPLC检测。3种寡糖标准品与样品的衍生化处理方法保持一致。HPLC检测系统是配有荧光检测器的安捷伦1260高效液相色谱系统,液相色谱柱为 HALO Glycan(4.6×150 mm,2.7 μm);荧光检测(FLD)激发波长Ex = 313 nm,发射波长Em = 358 nm;流动相A为0.05 mol /L甲酸铵水溶液,流动相C为纯乙腈;洗脱条件:0-2 min 80% C,2-10 min 80% C到55% C,10-12 min 55% C,12-12.5 min 55%C到80% C,12.5-15 min 80% C;流速为0.6 mL/min;进样量为3 mL。LC-MS检测系统是安捷伦6500系列四极杆(Q-TOF)液质联用系统,色谱条件同上,ESI负离子源,分子量扫描范围100-1 100。
1.2.5 工程菌株的优化 透酶的过表达:透酶基因lacY和nanT合成后,利用相同的载体构建方法,将lacY-nanT片段插入pUC18质粒得到pUC18-neuA-cst-lacY-nanT。将新的重组载体分别替换原菌株中pUC18-neuA-cst质粒,得到透酶过表达的寡糖合成菌株。启动子的更换:大肠杆菌启动子文库由武汉大学刘天罡教授馈赠,可以用于基因表达量的精确调控[19]。在大肠杆菌启动子文库中选取强度不同的启动子,以pUC18质粒原Lac启动子强度作为参照,实验中所用启动子由强到弱的排列顺序为pXET11、pXET31、pTrc、pLac、PXETa34、pXET41。在透酶过表达菌株的基础上,将5组启动子片段分别替换pUC18质粒上原有启动子片段,载体和菌株的构建方法同上。包含不同启动子的菌株进行平行发酵,以原菌株为对照,检测寡糖产量的变化情况。
2 结果
2.1 重组质粒及工程菌株的构建
扩增得到的neuA-cst片段的大小为1 551 bp,gne-cgtA片段的大小为2 158 bp,cgtB基因片段的大小为912 bp,neuA-cst-lacY-nanT片段大小为4 315 bp,与理论大小均一致。重组质粒pUC18-neuA-cst、pBBR1MCS3-gne-cgtA、pACYC184-cgtB和 pUC18-neuA-cst-lacY-nanT经基因测序验证正确,重组质粒pUC18-neuA-cst-lacY-nanT的PCR验证结果如图3-E。Strain-GM1、Strain-GM2、Strain-GM3的构建示意图如图3-A,3组菌株进行菌落PCR鉴定(图3-B-D),阳性克隆片段的大小均与理论大小一致,说明菌株构建成功。
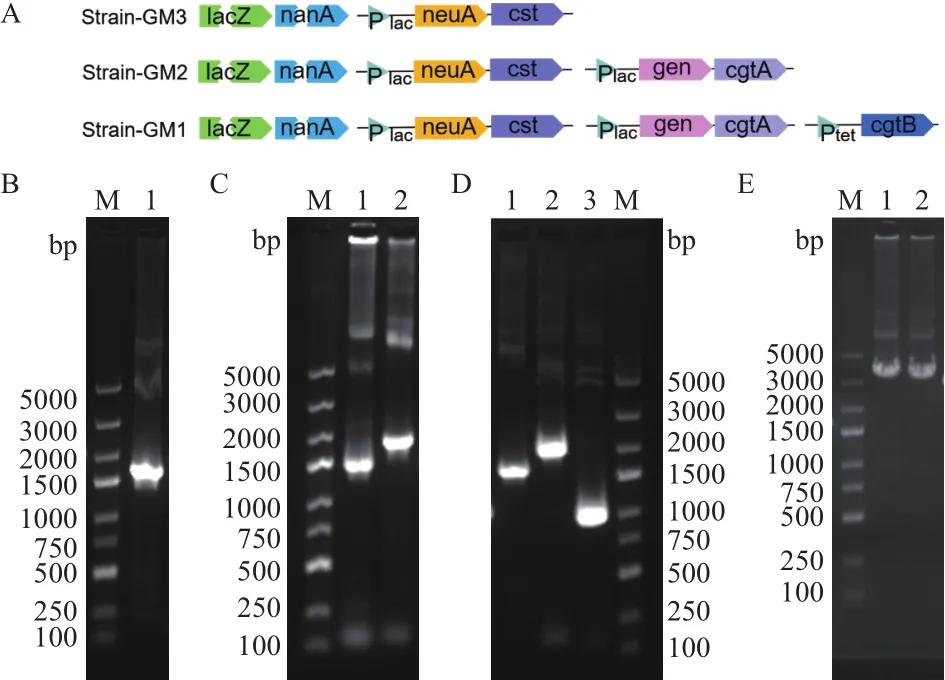
A:Strain-GM3、Strain-GM2、Strain-GM1 菌株构建示意图(断裂箭头表示敲除基因,实心箭头表示引入基因,三角形表示启动子)。B:Strain-GM3菌液PCR验证 ,M:DNA Marker;1:neuA-cst片段PCR产物。C:Strain-GM2菌液PCR验证, M:DNA Marker;1:neuA-cst片段PCR产物;2:gne-cgtA片段PCR产物。D:Strain-GM1菌液PCR验证, M:DNA Marker;1:neuA-cst片段PCR产物;2:gne-cgtA片段PCR产物;3:cgtB 片段PCR产物。E:质粒pUC18-neuA-cst-lacY-nanT的PCR验证,M:DNA Marker;1-2:neuA-cst-lacY-nanT片段PCR产物
2.2 寡糖定量检测方法的建立
利用高效液相色谱建立了GM3、GM2、GM1氟化寡糖的定量检测方法。3种产物检出限均小于0.1 mg/L,GM3、GM2、GM1 氟化寡糖标准品的保留时间分别为11.21 min、11.82 min、12.38 min。在0.5-100 mg/L检测范围内,质量浓度和色谱峰面积呈现良好的线性关系。工程菌株发酵液的检测结果如图4,LC-MS检测显示产物峰的m/z为634.2009、837.2798、999.3319,确定产物峰对应的化合物为GM3、GM2、GM1氟 化 寡 糖。Strain-GM3、Strain-GM2、Strain-GM1菌株发酵液分别在11.24 min、11.83 min、12.39 min出现目标产物峰,与对应标准品的保留时间一致。
2.3 工程菌株的发酵
Strain-GM3、Strain-GM2、Strain-GM1进 行 摇瓶发酵。取样时间为0、4、10、16、20、26、32、48、54、66、72、77和90 h。测定菌体的生长密度(OD600)并根据对应的标准曲线计算样品的浓度,菌株的生长及产量变化曲线如图5,3种菌株在0-26 h期间均快速增长,26 h之后均逐渐趋于稳定。GM3氟化寡糖在72 h左右产量最高达到约54.2 mg/L,GM2氟化寡糖在66 h左右产量最高达到约12.3 mg/L,GM1氟化寡糖在54 h左右产量最高达到约7.7 mg/L。
2.4 工程菌株的初步优化
外源底物氟化乳糖和唾液酸分别在乳糖透酶lacY和唾液酸透酶nanT的作用下进入细胞内,透酶过表达菌株的寡糖产量变化如图6-A。以原菌株作对照,3种寡糖的产量均有20%左右的提升。不同启动子强度的菌株平行发酵结果如图6-B-D)所示。以原Lac启动子作对照,GM3氟化寡糖合成菌株在利用pXET31启动子时产量提高约25%(图6-B),GM2氟化寡糖合成菌株最适合的启动子仍为Lac,启动子更换的菌株产量均没有提高(图6-C),GM1氟化寡糖合成菌株在利用pXETa34启动子时产量增长约28%(图6-D)。经过优化,GM3、GM2、GM1氟化寡糖的产量分别达到81.5 mg/L、18.1 mg/L和12.3 mg/L。
3 讨论
本研究通过在Escherichia coliJM107中异源表达CMP-NeuAc合成酶,UDP-GlcNAc异构酶和3种糖基转移酶cst、cgtA和cgtB,在大肠杆菌体内建立起神经节苷脂GM3、GM2和GM1氟化寡糖的生物合成途径。通过在大肠杆菌中表达两种底物透酶lacY和nanT来提高底物在细胞内的浓度,可以有效促进产物的生成。工程菌株启动子的优化显示,3种产物最适合的启动子强度均不相同。经过初步改造,3种寡糖的产量均有所提升,但目前产量仍较低且两种底物利用率不高,因此仍需要对人工合成途径进行更深入的解析和完善。
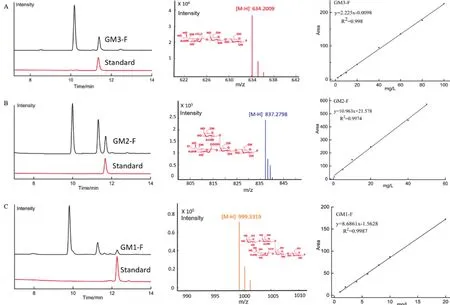
图4 工程菌株发酵液HPLC检测及LC-MS检测结果
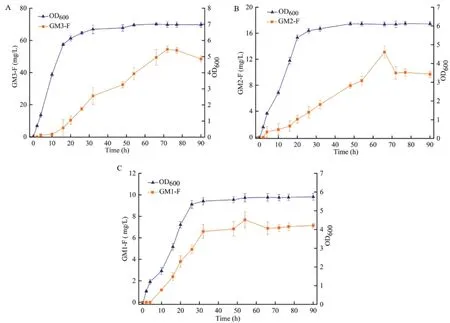
图5 Strain-GM3(A)、Strain-GM2(B)、Strain-GM1(C)菌株生长及产量曲线
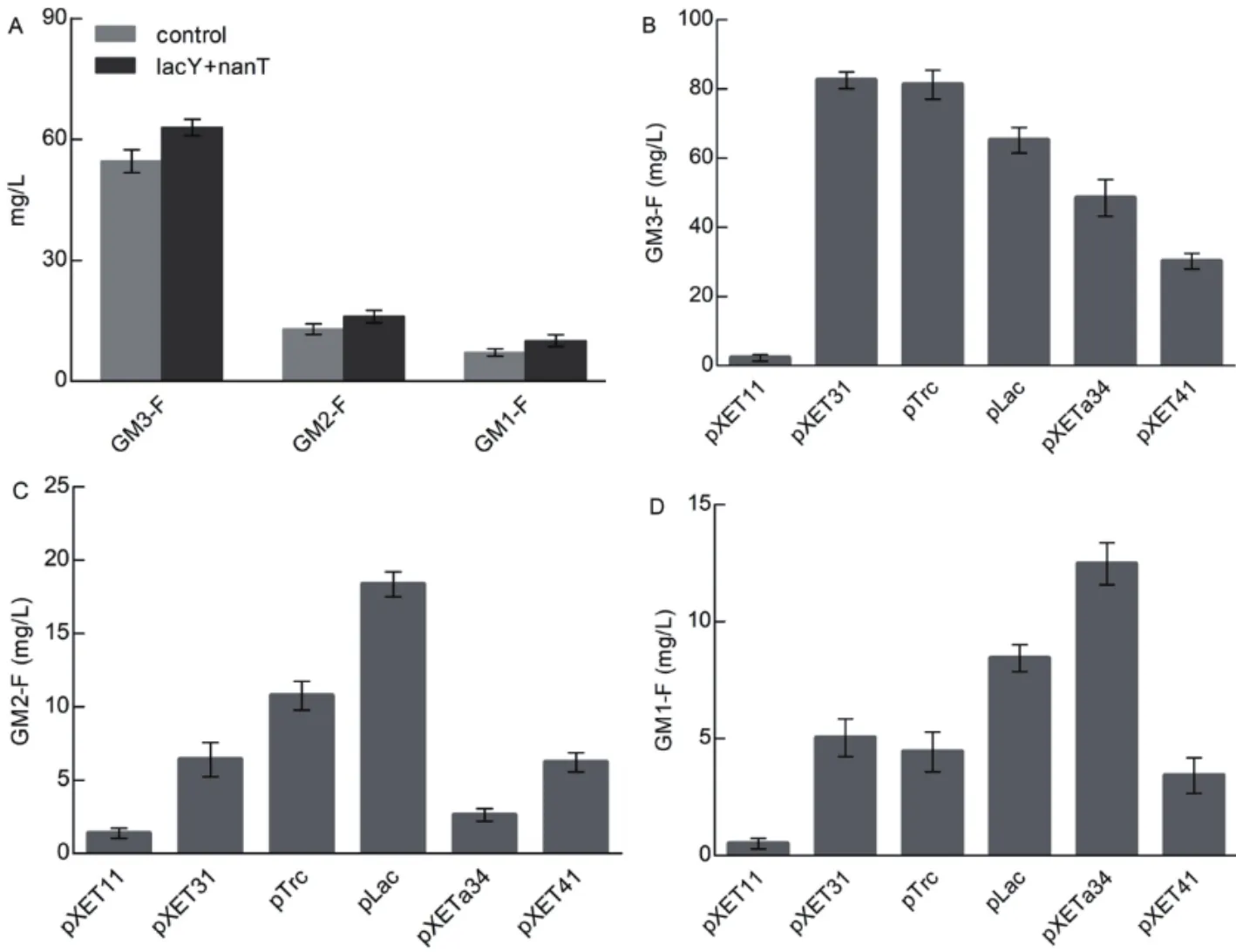
图6 工程菌株的初步优化
随着代谢途径的延长,GM3、GM2、GM1氟化寡糖的产量呈明显下降趋势,说明在代谢途径中可能存在消耗寡糖产物的旁路途径,或是外源表达的糖基转移酶活性不足从而限制了下游产物的生成。使用更高效的同工酶有望进一步提高寡糖的产量,如近来Klaudia等[20]发现并表征来源于Bibersteinia trehalosiand的一种新的α 2,3-唾液酸转移酶(BtST1),该酶在以乳糖为底物时,其催化活性是已报道同类酶中最高的,从而有利于唾液酸寡糖的合成。此外,也可以对代谢网络中各节点的中间代谢物建立系统的代谢组学分析方法,找到代谢途径中的限速步骤,为合成途径的优化提供指导。如武汉大学刘天罡团队在利用链霉菌进行多杀菌素的异源合成时,通过代谢组学的方法确定了多个多杀菌素链霉菌异源合成途径中的限速步骤,经过优化后的产量相比于出发菌提高了约1 000倍[21]。
4 结论
本研究通过在Escherichia coliJM107中构建神经节苷脂GM3、GM2、GM1氟化寡糖的生物合成途径。以氟化乳糖和唾液酸为底物,首次实现了3种神经节苷脂GM3、GM2、GM1氟化寡糖在大肠杆菌中的生物合成。
猜你喜欢
乳业科学与技术(2022年2期)2022-04-15
材料工程(2022年3期)2022-03-20
食品工业科技(2019年5期)2019-04-01
中国化肥信息(2018年6期)2018-08-23
中成药(2017年8期)2017-11-22
中国卫生标准管理(2015年17期)2016-01-20
中国当代医药(2015年33期)2015-03-01
中国当代医药(2015年31期)2015-03-01
中国当代医药(2015年9期)2015-03-01
中国塑料(2014年12期)2014-10-17
