
萃取-分光光度法测定铪中磷量
2019-08-02 09:55:06魏东翟通德李佗刘厚勇
四川有色金属 2019年2期
魏东,翟通德,李佗,刘厚勇
(西安汉唐分析检测有限公司, 陕西西安 710016)
铪具有可塑性、易加工、耐高温抗腐蚀的特点,是原子能工业的重要材料[1]。铪的热中子捕获截面大,是较理想的中子吸收体,可作为原子反应堆的控制棒和保护装置[2-4]。铪粉可作火箭的推进器,在电器工业上可制造X射线管的阴极。铪合金可作火箭喷嘴和滑翔式重返大气层的飞行器的前沿保护层。此外,在耐热合金如钨、钼、钽中,铪可作为硬质合金添加剂,提高合金的熔点[4]。
铪中多种杂质元素的测定有报道[5-7],磷的测定未见报道。磷是铪合金中的有害元素,标准[8]中规定了原子能级的HHf-01中磷含量小于0.002%。磷的准确测定对海绵铪、结晶铪的生产、研制和应用等有极其重要的作用。
1 试验部分
1.1 仪器和试剂
(1)仪器:可见分光光度计,波长范围340nm~1000nm,杂散光≤0.5%。
(2)试剂:氢氟酸(ρ1.13g/L),硝酸(1+1):将硝酸(ρ1.42g/L),沸3min~5min,驱尽氮的氧化物后配制。硼酸饱和溶液,高锰酸钾溶液(40g/L),亚硝酸钠溶液(100g/L),钼酸铵溶液(100 g/L),正丁醇-三氯甲烷混合液(1+3),盐酸(ρ1.19g/mL)。金属铪(Hf≥99.95%)。
氯化亚锡溶液(10 g/L):称取1g氯化亚锡,置于干燥烧杯中,加入8ml盐酸(ρ1.19g/mL)溶解,用水稀释至100mL,混匀,用时现配。
磷标准贮存溶液:称取0.4394g预先在105℃~110℃烘干2h的磷酸二氢钾(优级纯),置于250mL烧杯中,用水溶解,移入1000mL容量瓶中,用水稀释至刻度,混匀。此溶液每毫升含100ug磷。
磷标准溶液(10μg/mL):移取10.00mL磷标准储存溶液,置于100mL容量瓶中,用水稀释至刻度,混匀。此溶液每毫升含10μg磷。
在分析中使用优级纯试剂和实验室二级水。
1.2 样品分析
称取1g试料,精确至0.001g,将试料置于铂皿中,加入10mL水,分次滴加约4mL氢氟酸溶解试料。滴加2~3滴硝酸加热至完全溶解。冷却后加入15mL饱和硼酸溶液,加入4mL(1+1)硝酸,滴加高锰酸钾溶液至试液呈稳定的紫红色,并过量3~5滴,加热煮沸2~3分钟,滴加亚硝酸钠溶液至紫红色消失,继续煮沸3~5分钟,驱尽氮的氧化物。取下冷却至室温,移入100mL分液漏斗中,使溶液体积为45mL,加入25mL正丁醇-三氯甲烷混合溶液,6mL钼酸铵溶液,振荡一分钟,静置分层。将有机相放入另一个100mL分液漏斗中,于水相中加入10mL正丁醇-三氯甲烷混合溶液再萃取一次,将两次萃取的有机相合并。于有机相中加入15.00mL氯化亚锡溶液,振荡15秒,静置分层,弃去有机相;将部分水相移入1cm比色皿中,以随同试样空白为参比,于分光光度计700nm处测量其吸光度,从工作曲线上查得相应的磷量。
1.3 工作曲线的绘制
移取0.00mL、0.20mL、0.40mL、0.60mL、0.80 mL、1.00mL磷标准溶液配制工作曲线A和0.00mL、1.00mL、2.00mL、3.00mL、3.00mL、4.00mL、5.00mL磷标准溶液配制工作曲线B,分别置于一组100mL铂皿中,加入4mL氢氟酸,滴加2~3滴硝酸,加入15mL饱和硼酸溶液,加入4mL(1+1)硝酸,放置5min~10min后移入100mL分液漏斗中,控制溶液体积为45mL。以下步骤同试样分析步骤,以试剂空白为参比,测量其吸光度,以磷含量为横坐标,吸光度为纵坐标绘制工作曲线。
2 结果与讨论
2.1 工作曲线与线性方程
分别配制工作曲线A和工作曲线B,按照分析步骤操作,以试剂空白为参比,于分光光度计波长700nm处和分光光度计600nm处分别测量工作曲线A吸光度和工作曲线B吸光度。以磷含量为横坐标,吸光度为纵坐标绘制工作曲线,得到线性方程和相关系数,结果见表1。

表1 工作曲线和线性方程
磷钼蓝对分光光度计波长范围600nm~700nm的单色光均有较强的吸收效果,波长700nm附近吸收效果最强,波长600nm附近吸收弱。可根据磷含量的高低,选取适宜的吸收波长均能准确测定磷含量。
2.2 溶解酸用量
准确称取1g试样(厚度不大于1mm的碎屑)于100mL铂皿中,分次加入不同体积的氢氟酸,试验结果见表2。
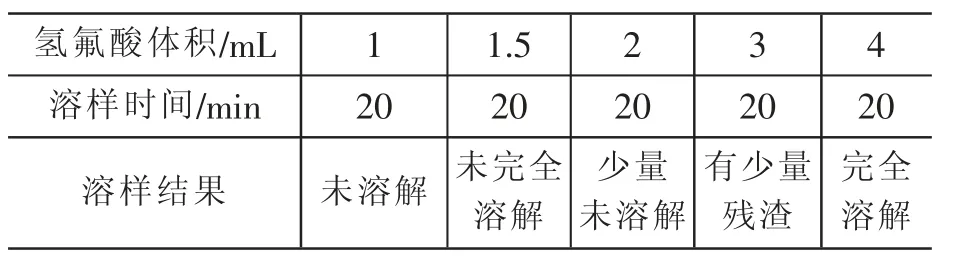
表2 溶解酸用量试验
由表2结果可知,溶样时加入4mL氢氟酸较为适宜。
2.3 显色酸度的确定
分取含磷5ug的标准溶液于一组分液漏斗中,分别加入1、2、3、4、5、6、7、8、9、10ml硝酸(1+1),按试验方法萃取,加入15mL氯化亚锡溶液反萃取。以试剂空白为参比,于分光光度计700nm处测量其吸光度,考察硝酸加入量对磷显色的影响,见表3。

表3 硝酸加入量对磷显色的影响
从表3可以看出,当硝酸的加入量为4mL~8mL时,样品吸光度较大且稳定,因此本实验选择硝酸的加入量为4mL。
2.4 基体铪的影响
分别移取0mL、0.20mL、0.40mL、0.60mL、0.80mL、1.00mL两组磷标准溶液,分别置于100mL铂皿中,于其中一组分别称取1g不含磷的金属铪于铂皿中。加入4 mL氢氟酸,滴加2~3滴硝酸,溶解后摇匀。加入15mL饱和硼酸溶液,加入4mL(1+1)硝酸,摇匀,静置5min~10min后移入100mL分液漏斗中,控制溶液体积约为45mL。以下按照显色萃取步骤操作。以试剂空白为参比,于分光光度计700nm处测量其吸光度,考察基体铪对磷显色的影响,见表4。

表4 基体铪对磷显色的影响
从上表比较可以看出,基体铪的加入对不同磷含量显色均无影响,不干扰磷的显色。

表5 萃取次数试验
2.4 萃取次数
移取1.00 mL、2.00mL、4.00mL磷标准溶液于两组铂皿中,称取1g不含磷的金属铪,溶解试样后,按照样品分析步骤进行试验,分别用正丁醇-三氯甲烷萃取一次和两次,测定磷量,试验结果见表5。
由表5可以看出,经过一次萃取,萃取率均能达到90%以上;经过两次萃取,萃取率均能达到96%以上。为保证萃取率,试验选择采用两次萃取。
2.5 共存元素的干扰
分别按照标准所载明的单一杂质元素和混合杂质元素的最高含量,考察其对5ug、10ug磷测定的干扰情况。
试验结果表明,30mg锆;800ug铁、镁;500ug铝、铬、钛、硅、氯;200ug铌、钽、钨均不干扰磷的测定。
2.6 精密度试验
目前只能提供纯金属铪,其磷含量小于0.0005%,无法形成含量梯度。试验以纯金属铪为基体,分别加入一定量的磷标准溶液制备模拟样(模拟样1#、模拟样2#、模拟样3#、模拟样4#),采用拟定的分析方法进行11次独立测定,计算平均值及相对标准偏差,结果见表6。

表6 精密度试验结果
从表6可以看出,对于纯金属铪中含磷量0.0004%~0.020%的样品,本方法的相对标准偏差均小于5.00%,说明该方法精密度好,测定结果准确、可靠。
3 结论
采用氢氟酸溶解样品,用饱和硼酸络合氟离子,在酸性介质中,磷酸离子与钼酸铵形成磷钼杂多酸,用正丁醇-三氯甲烷萃取分离,再以氯化亚锡还原成钼蓝并反萃取至水相中,以分光光度法测定磷的含量。在优化的条件下,本方法应用于金属铪样品中磷量的测定,测定范围0.0004%~0.020%,方法的RSD(n=11)均小于5.00%,具有较高的准确性和良好的稳定性。
猜你喜欢
云南化工(2021年9期)2021-12-21 07:43:44
云南化工(2021年6期)2021-12-21 07:31:26
供水技术(2021年3期)2021-08-13 09:08:36
电镀与环保(2018年1期)2018-04-04 05:21:22
浙江化工(2018年1期)2018-02-03 02:12:55
当代化工研究(2016年5期)2016-03-20 16:21:33
云南中医学院学报(2014年5期)2014-07-31 18:00:00
河南科技(2014年4期)2014-02-27 14:07:26
化学分析计量(2013年5期)2013-03-11 16:37:48
云南中医学院学报(2012年3期)2012-07-31 18:25:00
