
Cancer stem cells, stemness markers and selected drug targeting: metastatic colorectal cancer and cyclooxygenase-2/prostaglandin E2 connection to WNT as a model system
2019-07-30 14:13:44ReemAliALHulaisStephenJohnRalph
Reem Ali ALHulais, Stephen John Ralph
School of Medical Sciences, Griffith University, Menzies Health Institute Queensland, Gold Coast, QLD 4222, Australia.
Abstract Few studies have reported on the analyses of drugs targeting enriched populations of cancer stem cells (CSCs) as a means for identifying potent anti-CSC agents. This review evaluates recent information on the identification and functions of specific CSC surface markers, with particular emphasis on colorectal cancers and the screening of drugs to eliminate such cells. Many of these CSC markers are found commonly expressed on CSCs from different cancer types as well as embryonic stem cells. These markers are often related to hypoxic activation of the WNT/b-catenin pathway, cyclooxygenase-2/prostaglandin E signalling and their relationship to LGR5. By effectively using drugs that inhibit these pathways to kill the CSC population, or otherwise forcing them out of dormancy into active cell division, cancers should become more susceptible to chemotherapy. Such combinational therapies targeting both CSCs and proliferating tumor cells should greatly improve upon the current basis for treatment.
Keywords: Cancer stem cells, colorectal cancer, markers, selective drug targeting
GENERAL BACKGROUND TO CANCER STEM CELLS
Our understanding of the roles played by cancer stem cells (CSCs), their importance during the progression of cancer and justification for why they should be specifically targeted to eliminate cancer as a disease remains limited. Evidence supporting the “cancer stem cell” hypothesis is mounting such that CSC existence has become generally accepted. Analysis of PubMED reveals the increasing importance of CSCs with over two thousand publications in 2008 increasing to nearly 7000 annually at the present time and 70,831 in total. The involvement of the CSC population and their contribution to the circulating tumor cells (CTCs)and metastatic cancers is also becoming established. After therapy, drug resistant and dormant CSCs reactivate as major contributors towards tumor recurrence, generating further tumor masses. From this latter perspective we propose that it will be essential to not only identify the CSCs, but also the means for selectively targeting these cells to eliminate them if we are to improve upon current cancer therapy. In recent years, several reviews have covered the identification of markers for colorectal CSC (CR-CSC)[1], including that of the definitive colorectal cancer (CRC) stem cell marker, LGR5[2-4].
In 2010, the question was posed: “How can we identify and analyze colon CSCs and what agents are being designed to kill this chemotherapy-refractory population?”[5]. The CSC model accounts for tumor initiation,metastasis, drug resistance, and relapse and CSCs within the tumor bulk have the capacity to self-renew,differentiate, and give rise to new tumors (reviewed in[6-8]). In the present systematic overview, the focus is the identification and drug targeting of CSC populations and developing strategies for improving the therapy of advanced stage colorectal cancer by eliminating CSCs. Controversy has grown around the proposal of targeting the CSC population and whether it would be beneficial to patient outcomes[6]. In their treatise on the problems with the CSC model, these authors pointed out tumor heterogeneity and growth dynamics,which can be interpreted as cell plasticity in that cancer cells in a more differentiated state can re-enter the CSC pool. In addition, they argue that targeting a rare population of tumorigenic cells (such as CSCs)without consideration of the bulk of proliferating cells may not change patient outcomes[6]. By necessity, the principle of clonal extinction applies in terms of the need to eliminate all of the cancer cells, including the CSCs and their differentiated progenitors if we are to overcome cancer[7]. Simply put, one must kill the roots of the tree, or it will grow back [Figure 1] and was originally proposed as the “Dandelion hypothesis” for CSCs[8]. This will require not only killing the rapidly dividing, more differentiated cancer cell population as is currently targeted by commonly used chemotherapies, but also the more dormant and drug resistant CSCs existing within the tumor population.
One of the key aspects of CSCs that should be borne in mind is their property of “stemness” and by this is meant not only their capability of self-renewal and differentiation, but also that despite being sourced from different tissue origins, the various types of CSCs with their less differentiated phenotypes, will share properties and markers in common with embryonic stem cells (ESCs), reflecting their similarity in terms of earlier stages of development[9,10]. It is only when the CSCs differentiate that they will express greater sets of uniquely characteristic markers for the particular cell lineages that they encompass[11]. It is in this light that the markers for the CR-CSCs are discussed below, and are discussed in reference to related findings of similar markers found expressed on the ESCs and CSCs alike across a range of different cancer types[9,10,12].
CSC IDENTIFICATION AND GENERAL DEFINITION
The definition of CSCs is similar to that applied to the normal tissue stem cells: the ability of a small subpopulation of cells existing within a tissue which when isolated and reintroduced into the host have the capacity to reform complete tissues containing the range of cellular phenotypes similar to the original tissue from whence the stem cells were derived[12,13]. Thus, CSCs possess characteristics associated with normal stem cells, specifically mulitpotency in the ability to give rise to all cell types found upon pathological examination of an excised tumor[11]. The most commonly used method for identifying and isolating CSCs is via their characteristic cell surface markers, many of which were originally discovered from studies of normal tissue development, including from hematopoiesis and embryonic stem cells and which are also found co-expressed on CSCs[9].
The major surface markers that have been used to identify colorectal CSCs to enable their definitive isolation are the focus of this review, as well as improved means for testing anticancer cytotoxic drugs targeting their elimination. These findings should then be applicable to other CSC types. Historically, CSCs were extracted from solid tumors and partially purified as a poorly or negatively staining side population (SP)by flow cytometry, so-called because of their hallmark characteristic to exclude the nuclear DNA staining,fluorescent Hoechst series of dyes, such that CSCs could be separated from the bulk of other tumor cells that were highly positive for the nuclear DNA stain[14]. These poorly stained “SP” CSCs often showed much greater stem-like self-renewal and increased tumor-initiating capability and were responsible for tumor recurrence because they were more resistant to many forms of chemotherapy[14].
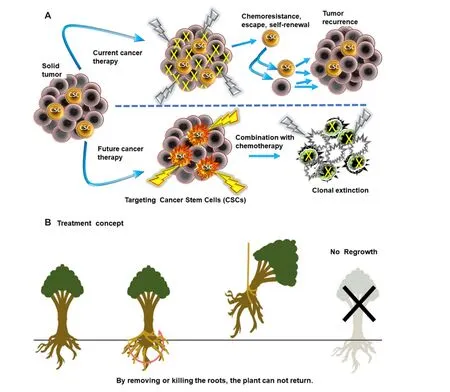
Figure 1. A: Targeting cancer stem cells: current treatments with conventional chemotherapy are not highly efficient against the cancer stem cells (CSCs) residing in tumors which resist chemotherapy or radiation and post-treatment will undergo self-renewal, differentiation and tumor regrowth or metastasize causing relapse and formation of additional tumors. However, drugs targeting the CSC population(such as celecoxib) can be used to eliminate the CSCs, either by direct cytotoxic effects or by sensitizing these tumor cells to other chemotherapy, resulting in the clonal extinction of the entire tumor cell population; B: The treatment concept of targeting CSCs (also known as the “Dandelion hypothesis”) by analogy proposes that by eliminating the roots (CSC population), the plant (tumor) can not become re-established whereas cutting the flowering stalks, stem or branches away will allow regrowth[8]
Other methods have also been used to enrich for CSCs, including growing tumor cells suspended in serumfree defined media or above agarose to form spheres or spheroids or by selective uptake of the AldefluorTMstem cell stains because CSCs show greater expression of aldehyde dehydrogenase 1[15]. The use of such methods for the identification and definition of CSCs has been complicated by observations that in the case of leukemias, only a few cancer cells isolated from the blood of leukemic animals were sufficient for transferring the leukemia into naïve healthy animals receiving the transfused cells[16]. Thus arose the basis for disputing the very existence of CSCs and whether all cancer cells are self-endowed with the capability of giving rise to whole tumors.
Since the discovery of CSCs, we have witnessed further complexities with the emergence of the epithelial to mesenchymal transition (EMT) hypothesis by which it was proposed that tumor cells transit back and forth between the two different states during the processes of invasion, migration and metastasis to form tumors at distant sites[17]. However, this view has been made less certain given recent evidence showing that multiple phenotypes are present within both the tumors and CTCs, which exist rather as mixtures of cells including both epithelial and mesenchymal types[18]. The emerging evidence from a range of studies would rather suggest a dynamic tumor heterogeneity with cancer cell plasticity existing as the consequence of genetic and metabolic changes, environmental differences and reversible adaptation of cellular properties, all proceeding within the context of a heirarchical system of tumorigenic CSCs differentiating into non-tumorigenic progeny (for reviews, see[19-21]).
During the course of progession within the primary tumor microenvironment, with expanding growth and increasing cellular demands on the limited availability of nutrients and oxygen supply, there follow frequent rounds of intermittent hypoxia, hypoglycemia and acidosis occurring within the tumor microenvironment[22]. Such conditions are the drivers for the production of greater numbers of the CSCs as a percentage of the total tumor population (reviewed in[23]). In addition, such stressors within tumors give rise to cancer cells showing a higher capacity to invade into nearby tissues, extending beyond the tumor boundary, migrating from the site of the primary tumors into the circulatory system to produce metastases.In this manner, the tumor spreads into distant locations, some involving advanced release of exosomes,interactions with immune cells and extracellular matrix to lay down the framework for future tumor beds at these remote sites as premetastatic niches[24].
Recent evidence has shown that the CTCs have a stem cell phenotype as evidenced by their expression of embryonic transcription factors (including OCT4, SOX2 and NANOG[25]), and contain mixtures of epithelial and mesenchymal phenotypes (reviewed in[26-28]). Furthermore, analyses of CTCs derived from the peripheral blood of colorectal cancer patients showed that they exhibit a more CSC-like phenotype[28], including expression of CSC markers such as LGR5, establishing these as prognostic markers for disease progression and metastasis in CRC patients. From these observations, it would appear that the CTC population includes CSCs in their mix and that such cells are highly metastastic [Figure 2]. Despite these complexities, our ability to identify and target the CSC populations with specific cytotoxic drugs must improve if we are to eliminate the cause of cancers and their metastasis, given their significantly enhanced tumor-initiating potential.
CR-CSCS, WNT SIGNALLING AND SURFACE MARKERS IN COMMON WITH OTHER CANCER
TYPES
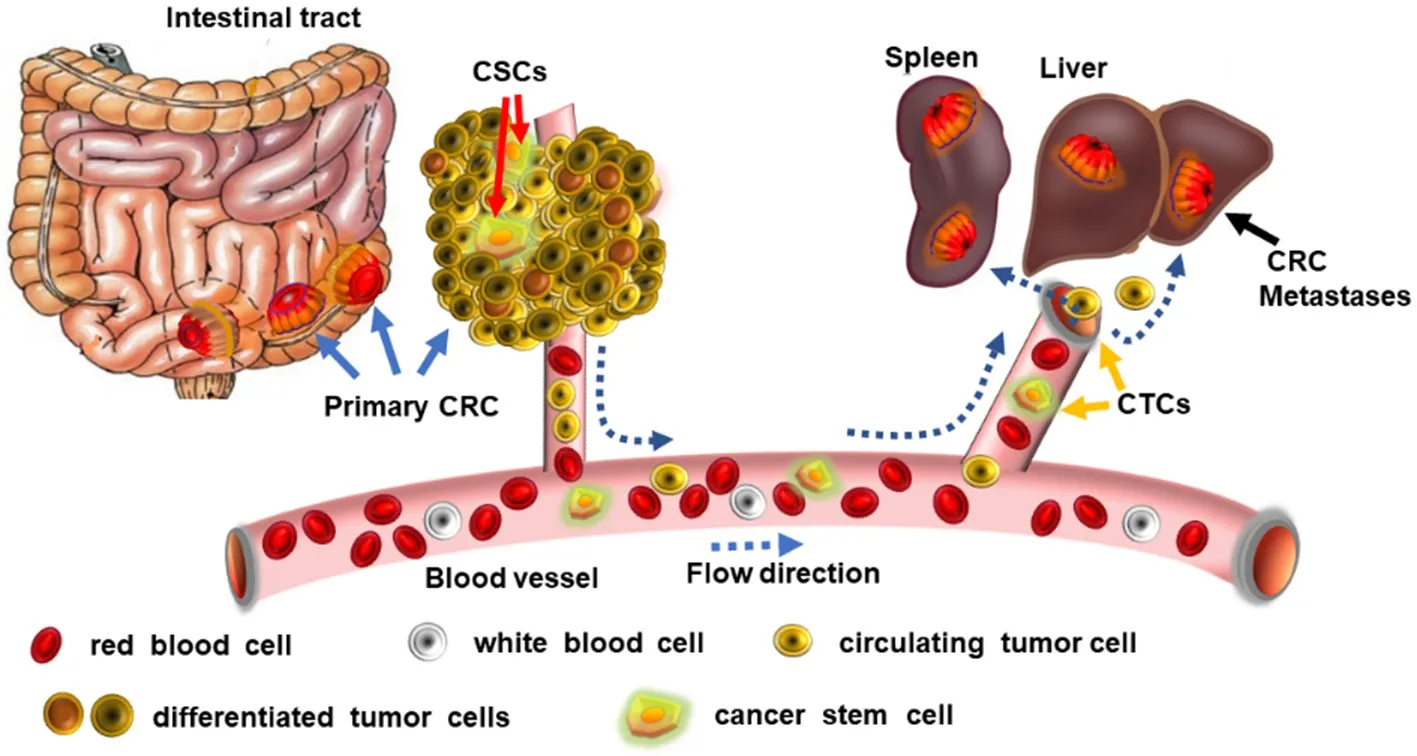
Figure 2. Circulating tumor cells (CTCs) include cancer stem cells (CSCs): tumor cells can migrate around the body via the blood vessels and this population includes CSCs that metastasize to invade into other organs, forming tumors by colonizing at distant sites such as in the spleen, and the liver. By applying drugs that are cytotoxic for CSCs (such as celecoxib) greatly reduces the incidence of tumor metastases and tumor recurrence. CRC: colorectal cancer
The self-renewal of CSCs is regulated by various modulators, including WNT/b-catenin, Notch and Hedgehog signalling, and at the transcriptional level by the pluripotency transcription factors such as OCT-3/4, KLF4,SOX2, and c-MYC, chromatin remodeling complexes, and non-coding RNAs (reviewed in[29]). WNT signalling cascades cross-talk via the fibroblast growth factor (FGF), Notch, Hedgehog and transforming growth factor beta (TGFb)/bone morphogenetic protein signalling cascades to regulate expression of the functional CSC surface markers that have been identified, such as CD44, CD133 (PROM1), epithelial cell adhesion molecule (EPCAM) and LGR5 (GPR49) (reviewed in[30]). The CSC regulatory signalling pathways such as WNT have become a target with implications for therapeutic interventions in cancers, albeit that its complexity poses significant challenges[31]. The WNT/b-catenin pathway is involved in the regulation of CSCs as with normal stem cells and their differentiation during embryogenesis and in the adult[30,31].The great majority (> 90%) of colorectal cancers have mutations in one of two genes involved in the WNT signalling pathway: either in the adenomatous polyposis coli or b-catenin (CTNNB1) genes and this has meant that WNT signalling has become an important cancer therapeutic target[32]. In 2016, studies examining the role of the Traf2- and Nck-interacting kinase (TNIK), an essential regulatory component of the WNT regulated T-cell factor-4 (TCF-4) and b-catenin transcriptional complex, showed that the small drug molecule NCB-0846 inhibited TNIK and abrogated colorectal cancer stemness[33]. This drug was potent at inhibiting the growth of colon cancer patient derived xenograft tumor models and resulted in smaller tumors. Hence, based on the above studies it is clear that WNT signalling plays a key role in the regulation of CR-CSCs. We ascertain that the relationship of the CSC markers to colorectal cancer provides a unique opportunity for anticancer drug discovery and on this basis the CR-CSC markers that have been identified to date are discussed further in the next sections. Their relationship to the WNT/b-catenin pathway for stem cell regulation is also highlighted where relevant.
The ATP binding cassette transporters/multiple drug resistance/P-glycoproteins
As described above, the ability to exclude the fluorescent nuclear DNA stains was an early hallmark of CSCs and has since been ascribed to their higher surface expression levels of the ATP binding cassette transporters(ABC) (a.k.a multidrug resistance or P-glycoproteins)[34]. These proteins are expressed on the outer cell membrane and will pump compounds such as chemotherapeutic drugs or DNA stains out of the cancer cells. One member of the family, ABCG2 (a.k.a. breast cancer resistance protein) was shown to expel a wide variety of exogenous and endogenous compounds from liver CSCs[35]and is prevalent on human colorectal cancer samples[36]. Membranous ABCG2 levels of expression in colorectal cancer independently correlate with shortened patient survival times[37].
The ABC transporters play significant roles in the distribution, absorption and elimination of substrate drugs and bestow multidrug resistance to cancer cells by maintaining the export or efflux of chemotherapeutic agents, preventing entry thereby avoiding attaining the toxic levels inside the cancer cells required to kill them. ABCG2 overexpression has also been observed in some human cancer cell lines and is widely expressed on liver cancer stem cells[38]. Consequently, the ABC transporters are viewed and regarded as possible universal biomarkers for stem cells, both normal and cancer stem cells alike[39]. Moreover, the evidence supports ABCG2 as playing a vital role in enhancing stem cell proliferation and in the case of esophageal squamous carcinoma has been shown to be required for the maintenance of the stem cell phenotype[39,40].
Therefore, ABCG2 has applications not only in the identification of the CSCs, but also for allowing the possibility of blocking its function as one way for improving selective drug targeting of tumors and their CSC population during the course of anticancer chemotherapy[41]. Despite the supportive evidence that ABCG2 is a stem cell marker, there are limitations since the exact role of the ABCG2 signalling pathway and its regulation is not well understood[42,43]. Although the molecular mechanisms regulating ABCG2 expression are not clearly understood, it is likely that the MYC oncogene plays an important role in promoting its expression in some cancers[44].
In studies of “SP” cells isolated from human colorectal cancer cell lines, it was shown that expression of ABC transporter genes, such as ABCB1 and ABCG2 was significantly higher, linked with greater resistance to 5-FU and irinotecan, and higher activation of the WNT signalling pathway, than for the non-SP cells[45]. In addition, silencing b-cateninexpression to inhibit WNT decreased significantly more SP cells than non-SP cells, decreased transcription of the ABC transporter genes and the silenced cells became relatively sensitive to paclitaxel and irinotecan. Hence, these results indicate that ABC expression is regulated by WNT signalling. EMT induces a switch in WNT signalling from a b-catenin/E-cadherin/Sox15 transcription factor complex to the b-catenin/Twist1/TCF-4 complex, the latter of which then binds to promoters of the CSC-related genes[46]. In this study, it was shown that Twist1 binding to b-catenin enhanced the transcriptional activity of the b-catenin/TCF-4 complex, including by binding to the proximal promoter region of theABCG2gene to promote ABCG2 expression as one CSC marker[46].
Zhanget al.[35]showed that the sensitivity of hepatocarcinoma cells to the chemotherapeutic drugs,doxorubicin or 5-fluorouracil inversely correlated with surface levels of the ABCG2 marker. The levels of ABCG2 expressed on cancer cells including colon cancer lines also closely correlated with tumorigenicity,drug resistance, proliferation and metastatic ability and therefore, the ABC transporters could be considered as critical and universal biomarkers for all CSCs[34,35,47,48]. The results above of WNT signalling correlating with ABCG2 expression levels also support strategies aimed at inhibiting the WNT signalling pathway as a means for targeting chemotherapy-resistant colon cancer cells, including the CSCs.
CD133
CD133 (a.k.a. prominin-1) has been widely touted to be a CSC marker and was originally recognized as a stem cell marker found on rat neuroepithelial stem cells[49]. CD133 and its role as a CSC biomarker has recently been reviewed[50]. As a common biomarker, CD133 has been used for identifying many different types of CSCs, including those originating from gliomas[51], colorectal[10,52,53], lung[54], liver[38], and prostate cancer[55]. However, CD133 has been shown to function in regulating glucose uptake for glucosamine production under conditions of increased glucose and glycolysis and is involved in autophagy[56,57].
Despite the fact that CD133 can mark the tumor-initiating cell populations in several solid tumors, studies have shown that it does not play a crucial role in the process of CSC maintenance. Thus, unlike knocking down CD44 expression which inhibited tumorigenesis of CR-CRC cells, knocking down CD133 gene expression had no effect[58]. CD133 expression is also not restricted to the stem cell population because both the CD133+and CD133-metastatic colon cancer cells were equally capable of initiating tumor production[59].Hence, CD133 expression is more a reflection of the level of glucose availability and is not necessarily a specific marker of CSCs.
CD44
CD44 is an extensively glycosylated surface protein as a major component of the extracellular matrix involved in cell-cell interactions, cell adhesion and migration and is one of the cellular receptors for hyaluronic acid (HA), thereby linking binding to selectin, collagen, osteopontin, fibronectin and laminin in the extracellular matrix[60]. HA binding to CD44 also facilitates complex activation of receptor protein tyrosine kinases of the epidermal growth factor receptor family in several types of cancer[61]. Upon HA binding to CD44 on the cell surface, increased levels of cell proliferation and survival occur through downstream signal activation of the MAPK and PI3K/AKT pathways[62]. Studies have also indicated that CD44 plays a significant role in the invasive and tumorigenic stemness capacity of several tumor cell types,including breast[63], prostate[64], pancreatic[65]and mesothelioma[66,67]. CD44 surface levels on tumor cells were shown to positively correlate with prostate cancer CTCs in the bloodstream of patients[68].
Genetic knockdown of CD44 prevented the formation of tumors by colorectal CSCs[58]. CD44 either alone or in combination with other cell surface markers can be used to enrich for CSCs from multiple tumor types including breast[69,70], prostate[71,72], colon[73,74], pancreas[65], and head and neck squamous cell carcinomas[75].However, given the relatively high levels of CD44 expressed on the cancer cell population, it would appear that CD44 is more a marker of the invasive, metastatic cell population and is not specific, per se for the CSCs.
CD13
The CD13 gene encodes the enzyme aminopeptidase N, a Zn2+dependent membrane-bound ectopeptidase preferentially degrading proteins and peptides with an N-terminal neutral amino acid. CD13 is overexpressed in multiple cancer types as well as on the surface of vasculature endothelial cells in tumors undergoing angiogenesis, making it a promising target[76]. CD13+cells form clusters or foci within the tumors, are typically in the G0phase of the cell cycle and also found present in the fractionated “SP”of CSCs from hepatocarcinoma[77,78]. Haraguchiet al.[79]showed that CD13+cancer cells surviving after chemotherapy or radiation treatment in tumors removed from hepatocarcinoma patients were found present within enriched fibrous capsular regions of the tumors and had decreased levels of DNA damage caused by reactive oxygen species (ROS). Thus, they proposed that the cancer cells appeared to be protected from treatment-induced apoptosis by expressing CD13. In their mouse xenograft models treated simultaneously with a CD13 inhibitor (either using a neutralizing antibody or the peptidase inhibitor, Ubenimex) and the genotoxic chemotherapeutic drug, 5-fluorouracil (5-FU), the combination diminished tumor levels significantly and was far more effective than 5-FU used alone[79]. Their studies have since been confirmed in other studies where CD13 inhibitors were used as an adjuvant therapy together with ROS-inducing chemotherapy or radiation treatment as a potential treatment method that could improve survival rates of patients with hepatocarcinoma[79-81]. The tripeptide NGR that targets CD13, conjugated with anticancer drugs has been similarly used to target human hepatocarcinoma growth by killing cancer stem cells and suppressing angiogenesis[82]. Although CD13 expression has been associated with human colon cancer and poorer prognosis[83], targeting its functional role as a marker on CR-CSCs has not yet been tested such that the usefulness of CD13 as a CR-CSC marker remains unclear.
LGR5
LGR5 is a member of the Leucine-rich-repeat-containing G-protein-coupled receptors (also known as GPR49) which belong to the seven transmembraneous G-protein-coupled receptor superfamily. The LGR1-5 family are regulatory receptors involved in WNT signalling[84]and can bind to the furin-like repeat FU2 domains of the R-spondin 1-4 stem cell growth factors to potentiate WNT signalling[85,86]. However,R-spondin 1 (RSPO1)/LGR5 can also directly activate TGFb signalling in a cooperative interaction with the TGFb type II receptor on colon cancer cells to enhance the TGFb-mediated growth inhibition and stress-induced apoptosis[87]. Knockdown ofLgr5attenuated downstream TGFb signaling and increased cell proliferation, survival, and metastasis in an orthotopic model of colon cancerin vivo[87]. Upon RSPO1 stimulation, LGR5 formed complexes with TGFb receptors[87]. Hence, the net effects of LGR5 expression will also depend on its interplay with both the WNT and TGFb signalling systems as well.LGR5 is found expressed in many organs including the brain, reproductive organs, mammary glands,the intestinal tract, stomach, hair follicles and the eyes[88]. TheLgr5gene is itself a WNT signalling target and prostaglandin E2 (PGE2) treating colorectal cancer cell lines enhances LGR5 expression whereasLgr5knockdown using siRNA inhibited the PGE2 survival response and induced cell death[84]. These results suggest that the PGE2 promotion of colorectal cancer cell survival at least in part proceeds via increasing LGR5 expression. Given the relationship of LGR5 with WNT signalling and PGE2 production by cyclooxygenase-2 (COX-2), it is likely that NSAIDs cause regression in familial adenomatous polyposis patients by interfering with the LGR5/WNT mechanism for promoting cancer stemness and cell survival.X-gal staining using the transgenicLgr5promoter driving b-galactosidase revealed that, following villus morphogenesis,Lgr5expression became restricted to dividing cells assembling in the intervillus region and within the distal small intestine[89].Lgr5deficiency is also known to cause premature Paneth cell differentiation within the small intestine, without affecting the differentiation of other cell lineages, nor the proliferation or migration of epithelial cells[89].
LGR5 was shown to be a biological marker for intestinal stem cells in the murine small bowel ofLgr5-EGFP-IREScreERT2 mice, as well as being useful for cell lineage tracing studies where the LGR5+stem cells developed into the different cell types comprising the intestinal crypt[88]. LGR5 expression has been closely linked with tumorigenesis including that of liver, colorectal, as well as ovarian cancers (for review,see[90]). In the normal intestine, LGR5 is solely expressed by the cycling crypt columnar cells and genetic lineage tracing studies identified these columnar cells in the base of the crypts as self-renewing, multipotent cells and are therefore considered to be genuine intestinal stem cells[91,92]. LGR5 has also been shown to be expressed in the stem cell niche by actively cycling cells of the murine hair follicle where lineage tracing and transplantation studies revealed that the LGR5+cells were retained for long periods and generated new hair follicles[93]. LGR5 expression in multiple other organs has led to the proposal that it represents a global marker of adult stem cells (reviewed in[94]).
Since LGR5 is involved in WNT signalling, it is not surprising that LGR5 and its ligands, the R-spondins are important for normal brain development[95]and hypoxia stimulates neural stem cell proliferation by increasing hypoxia-inducible factor-1alpha (HIF-1α) expression and activating WNT/b-catenin signaling[96]. Hence, it is likely that WNT acts by increasing LGR5 expression to promote stemness and brain CSCs[97]. LGR5 functions in the maintenance of brain CSCs and the transcribed levels ofLgr5is greater in these cells and in the case of glioblastoma has been associated with poorer prognosis[97]. In addition,immunofluorescence staining revealed the localization of LGR5+cells in sections of glioblastoma andLgr5siRNA knockdown led to lower expression of the neuronal L1 cell-cell adhesion molecule[97].
Most colorectal cancer cell lines and sporadic colonic adenomas exhibit significant LGR5 expression[98],although this is linked with stemness and renewal but not tumor progression because overexpressing LGR5 caused greater cell-cell adhesion, reduced tumor growth, invasiveness, migration and metastasis[98,99].Recently, LGR5 was established as a definitive surface marker for colorectal CSCs (reviewed in[90,100]),particularly when co-expressed with CD44 and EPCAM[101]. In these studies, the triple positive cells exhibited more pronounced CSC-like traits with LGR5-positive subpopulations showing higher capacities for colony formation, self-renewal, differentiation, and tumorigenicity as well as higher expression of stemness genes than did any other subpopulation. Thus, LGR5 is a marker for early CSCs and relates to their WNT promoted self-renewing capacity. Enhanced LGR5 expression remains persistent during adenoma to carcinoma transition in human CRC samples, but markedly declines in the budding cancer cells at the invasive tumor fronts, and was not associated with either WNT- or EMT-signalling pathways[98]. In the latter study, LGR5 overexpression attenuated proliferation, migration, and colony-forming capacities in colon cancer cells. Unfortunately, the levels of TGFb expressed by these cells were not analysed in these studies[96,98,99]given the observations above that TGFb switches the role of LGR5 to inhibit growth. These findings indicate that LGR5 can function as a tumor suppressor promoting maintenance of stemness during CRC progression and that LGR5 is a suitable marker of CSCs.
The most definitive studies for LGR5 as a CR-CSC marker were obtained by using a tamoxifen inducible suicide gene inserted into theLGR5gene of transgenic mice[2,4](reviewed in[102]). The suicide gene could be triggered upon expression of LGR5 and activated by the injection of tamoxifen, resulting in death of the colon CSCs by tamoxifen. The more differentiated tumor cells do not transcribe LGR5 and hence,do not express the suicide gene, such that they survived. When the suicide mechanism was activated in growing tumors in mice, the LGR5-positive CSCs were eliminated. As long as the suicide mechanism was maintained then tumors regressed but when their suicide was no longer induced (by tamoxifen withdrawal),then differentiated tumor cells filled the void and reacquired CSC qualities with rapid tumor regrowth.Furthermore, it was shown that metastasis and growth of preestablished metastases was halted. These findings showed that unlike cancer at the primary tumor site, metastases were dependent on preexisting LGR5+CSCs, so that once these CSCs were eliminated, reacquiring the CSC phenotype in the metastases was not possible[4]. Hence, LGR5 is expressed in CR-CSC and these studies support the Dandelion hypothesis[Figure 1B], such that advanced metastatic CRC should be amenable to elimination by anticancer drugs that target the CSCs.
CD326 (EPCAM)
The surface marker, EPCAM (also referred to as 17-1A, TROP-1, CD326, GA733-2, KS1/4 or gp40) is a type 1 transmembrane glycoprotein and a calcium-independent homophilic cell adhesion receptor of 30-40 kDa,encoded by theTACSTD1gene[103]. EPCAM is expressed by several human epithelial tissues, progenitor cells,cancer cells, stem and germ cells[104]. This protein marker was discovered initially as an antigen expressed on colon cancers that was involved in cell-cell adhesion and the structure of EPCAM comprises an extracellular domain, thyroglobulin domains, epidermal growth factor domains, transmembrane domain and a short intracellular domain (ICD) consisting of 26 amino acids known as EpICD[105,106].EpCAM (TACSTD1)may be an oncogene as the encoded EpICD can be proteolytically released to signal into the cell nucleus by engagement with elements of the WNT signalling pathway including activation of the b-catenin/MYC pathway resulting in tumor cell proliferation (reviewed in[106]). Expression of the EPCAM marker is elevated in malignant neoplasia and most cancers abundantly express EPCAM[104].
In normal human cells, EPCAM is located in the intercellular spaces at the tight junctions formed between adjacent epithelial cells[107]. Hence, it is inferred that EPCAM is sequestered in normal epithelial, whereas it becomes uniformly spread around the surfaces of cancer cells, where it is readily accessible to antibodies targeting it. The role of EPCAM in tumor progression and development is controversial, as is its relationship to another cell adhesion molecule, E-cadherin[105]. EPCAM has been proposed to act as a molecule for homophilic cell to cell adhesion, which would prevent the occurrence of metastasis[103]. It has a protective function in CRC since the deletion ofEpCAMleads to an increased risk of developing CRC[108]and when overexpressed on CRC cells, inhibits their metastasis[109].
In contrast, EPCAM expression inhibits the extent of E-cadherin mediated cell to cell adhesion, enhancing metastasis[110], although EPCAM has been proposed to bind to E-cadherin, both of which are important to the stability of the tight junctions (reviewed in[107]). In addition, overexpressing EPCAM in tumor cells can enhance WNT signaling to promote the proliferation of cancer cells[111]. In breast carcinoma, high levels of EPCAM expression are associated with poorly differentiated tumors[112], development of larger cancers,nodal metastasis and poor survival of the patients[113]. Marked EPCAM expression on breast carcinomas has been linked to poorer prognosis for both node positive and negative diseases[114]. In 2009, the European Union approved the EPCAM antibody, Catumaxomab, a bivalent monoclonal antibody which cross-links between T cells (via CD3 receptor binding) and EpCAM on tumor cells in order to promote the elimination of ascitic tumor cells. This therapy has contributed greatly to the management of breast cancer patients with malignant ascites and peritoneal carcinomatosis[115]. EPCAM is also expressed by almost all human adenocarcinomas, retinoblastoma, hepatocellular and squamous cell carcinomas[116].
EPCAM can be downregulated when cancer cells undergo EMT[117], during the dissemination of cancer cells into other tissues. Hence, detecting cancer cells in blood as CTCs using EPCAM as a marker is likely to miss the invasiveness/migration events of tumor cells. EPCAM is also expressed by the normal breast epithelial cells, although its role in the homeostasis of normal breast tissue is unclear[118]. In the human kidney 2 (HK-2) proximal cell line grown under hypoxic conditions, the activity of theEpCAMpromoter is increased two fold and in both HK-2 cells and primary kidney proximal cells, EPCAM protein expression is increased after hypoxia and reoxygenation[119]. The widely distributed expression of EPCAM on most cancer cells makes EPCAM unsuitable as a specific marker of CSCs.
CD271 (p75NTR)
CD271 was identified as the low-affinity nerve growth factor receptor (LNGFR), also known as the p75 neurotrophin receptor (p75NTR) belonging to the tumor necrosis factor receptor and low-affinity neurotrophin receptor superfamily and is a marker for cells of neural crest embryonic origin[120]. Previously,human CD271 was shown to be expressed on cells of the peripheral and central nervous system and was suggested to be involved in the survival, development as well as differentiation of neuronal cells[121]. CD271 or LNGFR is also found expressed on a wide range of other cell types including follicular dendritic cells,mesenchymal stem cells (MSCs), astrocytes, Schwann cells, neural crest stem cells, oligodendrocytes,sensory and autonomic neurons and mesenchymal cells[122].
Studies have shown that CD271 (LNGFR) is a predominant biomarker expressed by MSCs isolated from the bone marrow[123]. The fibroblastic colony forming units (CFU-F) in MSC populations could be established only by the CD271+cell fractionation, whereas no CFU-F’s were obtained with the CD271-cell population.CD271 is a highly specific marker expressed on the surface of freshly isolated bone marrow MSCs[124]. CD271 may not be a specific marker for all tissues, because as a marker it failed to isolate MSCs from umbilical cord blood[123].
CD271 was identified as a marker for the CSC population within human melanomas and its usefulness as a melanoma CSC marker has been well documented[121]and was shown to be crucial for maintaining the tumorigenicity and stem-like properties of melanoma cells[125]. CD271 has also been characterized as a stem cell marker for a small percent of cells in oesophageal squamous cell carcinoma[126,127]and was associated with CSCs[128]with high metastatic potential in CTCs from these cancer patients[129]. CD271 confers an invasive and metastatic phenotype to head and neck squamous cell carcinomas through the upregulation of the EMT transcription factor, Slug[130]which also promotes stemness. Thus, CD271 appears to be a marker of CSCs across a range of cancer types.
CD271 overexpression on melanoma cells suppressedin vitroactivation of melanoma specific cytotoxic T lymphocytes[131]. Moreover, the expression levels of PD-L1 and CD271 on the melanoma cells were both increased by IFNγ such that PD-L1 levels were related to those of CD271, and together with PD-L1 strongly and additively suppressed melanoma specific CTL activation[131]. CD271 expression on melanoma cells has also been linked to p53 activity, the DNA damage response and chemotherapeutic drug resistance as well as migratory properties to form metastases[132-134]. The higher expression levels of CD271 may determine specific properties of brain trophic metastatic melanoma cells[135,136].
Hypoxia induced activation of the HIF-1α and HIF-2α as well as their target genes enhances tumorigenicity,self-renewal, resistance to chemotherapeutic drugs and metastatic potential and is associated with increased surface levels of CD271 as a stem cell marker (reviewed in[137,138]). However, CD271 expression has also been reported to inversely correlate with hypoxia (HIF-1α expression) in skin samples from human melanomas analysed histologically at different stages of progression[139]and likely reflects a delay in its expression after hypoxic induction. CD271 was noted to be highly expressed on the cells in the dermal nests of the biopsied melanomas from human skin samples[139,140].
In this regard, hypoxia and HIF activation induceOct4gene expression, which will in turn activateCD271[141]. Thus, transfected gene expression of OCT4 caused melanoma cell dedifferentiation, acquiring features of CSCs such as multipotent differentiation capacity and expression of melanoma CSC markers,ABCB5 and CD271[141]. Mechanistically,Oct4-induced dedifferentiation was associated with increased expression of endogenous stemness factors, OCT4, NANOG and KLF4, and changes in global gene expression enriched for general transcription factors. The reverse study, applying RNAi-mediated knockdown ofOct4in the dedifferentiated melanoma cells, led to diminished CSC phenotypes[141].Moreover, OCT4 expression in melanoma cells was induced by hypoxia and its expression was detected in a sub-population of melanoma cells from clinical samples[141]. Overexpressing CD271 on human melanoma cell lines was shown to reduce their invasive and metastatic traits, whereas CD271-melanoma cells showed less adhesive, more highly proliferative and invasive traitsin vitroandin vivo[142]. Mechanistically, similar to EPCAM, it was found that the CD271 ICD regulated proliferation[143]. CD271, originally expressed in the epidermis of skin reconstructs, also disappeared when melanoma started to invade the dermis[142]. Thus,CD271 has been identified as a suitable CSC marker, at least in the case of melanoma.
CD35 (Cripto-1)
Mouse and human Cripto-1 proteins range in sizes from 24 to 36 kDa and are found in complexes associated with other proteins of between 14 to 60 kDa. Cripto-1 is glycosylated atN- andO-linked asparagine and serine residues respectively[144]. It is a member of the epidermal growth factor (EGF)-related family of growth factors, also known as the EGF-Cripto-1-FRL-1-cryptic (CFC) family. The EGF-CFC proteins are all associated with the cell surface and their structures including an amino terminal region, a modified EGF-like domain, a preserved CFC cysteine rich domain and a short hydrophobic COOH-terminus for attaching to the glycosylphosphatidylinositol (GPI) membrane anchor[144]. The soluble, biologically active form of Cripto-1 generates from the detachment of the GPI by GPI-phospholipase D such that Cripto-1 can act either as a soluble ligand or a cell-membrane anchored protein[144].
Cripto-1 was found highly expressed in a subpopulation (up to 60%) of human embryonal carcinoma cells which showed CSC-like properties, expressing OCT4, SOX2, and NANOG. Cripto-1 has a role in the maintenance of stem cells and their malignant progression[145]as a multifunctional modulator during embryogenesis, as well as being important for oncogenesis[144,146]. In the process of embryogenesis, Cripto-1 plays an important role in association with the TGFb ligand, Nodal[147]. Cripto-1 is highly expressed in many different human tumors, findings consistent with its role as a marker of the undifferentiated CSCs[148-150].
Cripto-1 is one of the master regulators of embryonic development along with the Notch/CSL, and WNT/b-catenin systems[151]. Hence, Cripto-1 is a surface marker for the identification of stem and quiescent cancer cells including the CSC phenotype in colorectal cancers and similar to the other CSC markers LGR5,EPCAM and CD271 described above, Cripto-1 expression is also up-regulated by hypoxia when HIF-1α binds to the hypoxia response elements[145,152]within the promoter of human or mouse Cripto-1 genes[153].Hypoxia can enhance production of beating cardiomyocytes and modulates the differentiation of mouse ESCs and Cripto-1 is required for the hypoxia-induced differentiation of mouse ESCs into cardiomyocytes,helping to regulate stem cell self-renewal and proliferation[153]. It follows that Cripto-1 could be a potential marker for sub-populations of tumor cells with stem-like properties.
CD15 (stage-specific embryonic antigen 1)
Stage-specific embryonic antigen 1 (SSEA1; also called CD15 or non-sialylated Lewis X antigen) refers to a carbohydrate based antigenic epitope (3-fucosyl-N-acetyl-lactosamine) synthesized by fucosyltransferases(FUT4, FUT9) and is linked with cell adhesion, differentiation and migration. This marker is associated with the proteoglycan, phosphacan (a.k.a. protein tyrosine phosphatase receptor-Z1; RPTPZ1; PTPRZ1) which regulates the proliferation, self-renewal and differentiation of stem cells[154]. RPTPZ1 is a receptor for the extracellular matrix protein, tenascin C recently linked with EMT, metastasis, recurrence and poor survival of CRC patients[155].
CD15/SSEA1 is expressed by murine embryonal stem cells, murine embryonic carcinoma cells, as well as human and murine germ cells[156]. SSEA1 has been widely used as a positive surface marker for undifferentiated normal stem cells from mice but not for human stem cells[39]. Thus, upon differentiation of the murine embryonal stem cells or embryonal carcinoma cells, expression of SSEA1 is down-regulated,whereas during human embryonal stem or carcinoma cell differentiation, SSEA1 expression is increased.The SSEA-1 marker is commonly used for identifying stem cells because it can be readily detected using the monoclonal antibody, MC-480, as a cell surface antigen involved in cell differentiation. This monoclonal antibody has also been commonly used as a standard reagent for detecting embryonic stem cells[157].
SSEA-1/CD15/FUT4 was found commonly overexpressed on metastatic CRC patients (43%) and was associated with lower levels of intratumoral CD3+and CD8+T cells, as well as poorer patient outcomes in terms of responses to treatment or progression-free survival (PFS) than those CRCs that were CD15/FUT4-low or negative[158]. Studies of brain development have shown that expression of CD15/SSEA1 is predominantly associated withO-mannose-linked glycans of the phosphacan/RPTPZ1[159]and high levels of RPTPZ1 expression have also been reported present in human CRC samples[160]. Hence, although SSEA-1 may be a putative CSC marker in the case of murine CRC, it is likely to show a more widespread expression on human CRC cells.
COX-2 AND CELECOXIB AS AN APPROVED DRUG FOR COLORECTAL CANCER, ALSO TARGETS CSCS, INCLUDING CR-CSCS
Celecoxib was approved nearly 20 years ago by the USFDA for the treatment and prevention of some forms of colorectal cancers, including familial adenomatous polyposis and sporadic colorectal adenoma.The anticancer effects of celecoxib were claimed due to its potential for inhibiting the enzyme, COX-2 in the CRC cells[23,161-163]. COX-2 (also known as prostaglandin-endoperoxide synthase 2) uses arachidonic acid to produce the prostaglandin H2 required to synthesize the prostanoids including thromboxane,prostaglandins such as PGE2 and prostacyclin. COX-2 is overexpressed in many cancer cell types, including CRC[164]when compared to levels in the corresponding normal cells. Moreover, COX-2 was recently detected in the CTC populations from CRC patients, where COX-2 expression was associated with those tumor CTCs positive for vimentin and Twist as two markers detected co-expressed at a higher frequency in patients with metastases compared to those without (72.0%vs. 42.8%)[165].
The role of COX-2/PGE in CRC and relationship to LGR5/WNT activation and survival of CSCs
The tumor cell inhibitory mechanism of celecoxib was proposed to involve counteracting the increased levels of COX-2 enzymatic function in colorectal cancer cells [Figure 3], thereby causing the suppression of cancer cell growth ([166], reviewed in[167]). As outlined above, one of the mechanisms of action by inhibiting COX-2 likely involves interfering with the WNT/PGE2 signaling pathways that would otherwise promote greater levels of LGR5 and cancer cell stemness. Among the tumor-sustaining prostaglandins produced by COX-2, PGE2 is associated with enhancing cancer cell survival, growth, migration, invasion, angiogenesis,and immunosuppression[168]and can act in an autocrine/paracrine manner to promote tumor growth and survival[169][Figure 3]. Inhibiting COX-2 with celecoxib would thereby block the growth stimulating effects derived from the COX-2 production of prostaglandins as cytokine-like factors stimulating growth, CSCs and metastases[170,171]. Levels of PGE2 correlate with colonic CSC markers (CD133, CD44, LGR5, and SOX2 messenger RNAs) in human colorectal carcinoma samples suggesting that it is a marker for CR-CSC[171].Thus, PGE2 induced CSC expansion by activating nuclear factor-kappa B (NF-κB) via the PGE2 receptor 4(EP4) and PI3K/MAPK signaling, promoting the formation of liver metastases by colorectal cancer cell lines injected into mice[171].
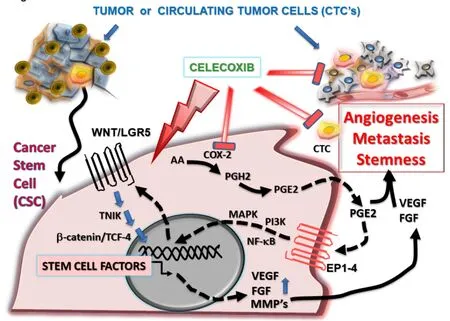
Figure 3. Celecoxib prevents CSC growth and tumor angiogenesis by inhibiting the COX-2/PGE2/VEGF and WNT/LGR5 stemness pathways. COX-2 is required for the production of prostanoids, including PGE2, which is released by tumor cells and stromal cells. PGE2 acts in an autocrine/paracrine manner binding to surface members of the prostanoid receptor family (EP1-4) to increase cancer cell stemness, angiogenesis (via production of VEGF and FGF), invasiveness (via matrix metalloproteinases MMP-2, MMP-9) and thereby promotes tumor metastasis. Celecoxib is an anticancer drug that is cytotoxic for CSCs, inhibiting COX-2/PGE2 production and stemness,thereby down-regulating the WNT/LGR5 signalling pathway, inhibiting angiogenesis and promoting the anticancer immune response.CSC: cancer stem cell; CTC: circulating tumor cell; VEGF: vascular endothelial growth factor; FGF: fibroblast growth factor; COX-2:Cyclooxygenase-2; PGE2: prostaglandin E2; PGH2: prostaglandin H2; TCF-4: T-cell factor-4; TNIK: Traf2- and Nck-interacting kinase; AA:arachidonic acid
Angiogenesis is one of the classical hallmarks of cancer, promoting new blood vessel formation from preexisting vessels, thereby facilitating the supply of blood borne nutrients and oxygen to the tumor cells[172].As early as 1999, celecoxib was recognized as an important chemotherapeutic agent in colorectal cancer treatment[173]because of its ability to potently inhibit angiogenesis by preventing vascular endothelial growth factor (VEGF) and FGF production, reducing growth of gastrointestinal cancer xenografts in nude mice[174]. COX-2/PGE2 activation increases VEGF expression by colon cancer cells, thereby promoting tumor angiogenesis (reviewed in[175,176]).
Evidence for COX-2/PGE playing a role in many other cancer types besides CRC and is a CSC drug target
These early findings have since been confirmed in many subsequent studies of other cancers as well including lowering microvessel density in gastric cancer models by inhibiting COX-2 mediated PGE2 production and VEGF expression[177,178], and in head and neck[179], pancreatic[180], Lewis lung cancers and sarcomas[181](reviewed in[182]). In more recent studies of dimethylhydrazine-induced colorectal cancer models, NSAIDs like celecoxib decreased the rate of adenocarcinomas, markedly lowering angiogenesis parameters including VEGF, MMP-2, MMP-9 and MCP-1 levels[174,183]. More importantly, inhibiting COX-2 with celecoxib was found to block the PGE2-induced tumor repopulating capacity, which otherwise promoted chemoresistance in bladder cancer xenografts[184]as well as CSC expansion and metastasis in colorectal cancer models[171].
The role of COX-2 in CSC survival and repopulation after therapy has been reviewed elsewhere[185], such that it has become clear that inhibiting COX-2 is an effective method for preventing treatment failure due to tumor repopulation. Moreover, celecoxib, as a selective COX-2 inhibitory drug, has also been reported to promote the apoptosis of colon cancer cells[186]. The prostaglandin, PGE2 produced by COX-2 activates prosurvival pathways preventing apoptosis of colon cancer cells[187](reviewed in[168,188]). Celecoxib, by inhibiting COX-2 facilitates the activation of various apoptotic pathways in colorectal cancer cells, including p53, p38 and BAX pathways[189].
Another aspect of the prostaglandins produced by COX-2 activity is that some, such as PGE2, are proinflammatory cytokine-like factors that modulate the anticancer immune response[190]. Celecoxib’s anticancer inhibitory effects (resulting from lower prostaglandin levels) also include promoting an enhanced immune response. Thus, celecoxib inhibition of COX-2 prevents the switching from a Th1 to a Th2 immune response, where Th2 would be more favorable to cancer cell survival. The immune response switch to Th2 is induced by increased levels of COX-2 which also leads to decreased production of the NF-κB activated cytokines that would otherwise induce Th1 proliferation, including interferon gamma (IFN-γ), tumour necrosis factor-alpha, and IL-2. Moreover, COX-2 activity promoted expression of cytokines including IL-6, IL-4, and IL-10 which preferentially activate Th2 type immune responses[191], thereby enabling the cancer cells to evade the host immune system.
Tumor-associated COX activity in a mouse melanoma model driven by oncogenic mutation inBraf(similar to the situation in human melanoma) was shown to be a key suppressor of type II IFN (IFN-γ) and T cellmediated tumor elimination, inducing an inflammatory signature more typically associated with cancer progression[192]. COX-dependent immune evasion was also shown to be critical for tumor growth in a range of models including melanoma, colorectal, and breast cancers[192]. Notably, tumor immune escape could be reversed by a combination of an immune checkpoint blockade inhibitor together with COX inhibitors.Hence, the COX-2/PGE pathway can promote tumor growth, survival and evasion of the immune response and highlights the importance of using NSAIDs such as celecoxib to inhibit the COX-2/PGE based activities in tumors.
We have shown that celecoxib has a fourth component to its anticancer function based on direct effects in killing cancer cells by targeting their mitochondria to increase the respiratory substrate driven production of reactive oxygen species such as superoxide, thereby activating the intrinsic apoptotic pathway[23,193]. Although many of the NSAIDs show similar properties and ability to act as cytotoxic anticancer drugs (reviewed in[194]), celecoxib was found to be by far the most outstanding[23,193]and was highly effective at inhibiting growth of triple negative breast cancer spheroids[195]. In this regard, in many animal models of cancer,celecoxib has been shown to chemosensitize cancer cells in a synergistic manner to enhance the cytotoxic effects of commonly used chemotherapies, such as the anthracycline drugs doxorubicin and epirubicin[196-200]and platinum-based chemotherapies[164,199,201-206]. In murine models of colorectal cancer, synergistic anticancer effects were obtained by combining celecoxib with the common CRC chemotherapeutic drug, 5-fluorouracil(5-FU)[207]or with oxaliplatin[208].
In human hepatomas, Chuet al.[209]showed that celecoxib promoted death of the CSC population and suppressed stemness and progression by up-regulating the tumor suppressor function of the tyrosine phosphatase, PTEN. In related studies[84,210], celecoxib was shown to target CSCs and suppress their selfrenewal, sensitizing them against chemoresistance as well as inhibiting their EMT. Further, these studies showed that celecoxib attenuated metastasis and tumorigenesis by inhibiting the synthesis of PGE2, thereby down-regulating the WNT/LGR5 pathway activity, including for colorectal CSCs[84]. Hence, celecoxib has shown a range of highly beneficial and intriguing properties as an effective anticancer drug in animal tumor models and provides an example of a drug affecting the WNT/LGR5 signaling pathway in CSCs to prevent CSC survival and metastasis.
Clinical studies of colorectal cancer patients receiving treatment with celecoxib over the long-term for several years have been successfully completed[211-214]. In most of these studies, celecoxib was used in combination with chemotherapy and showed significant improvements with inclusion of celecoxib, resulting in decreased rates of metastases and incidence of recurrence. In a large trial from 2006, the results of the Adenoma Prevention with Celecoxib (APC study) showed that continued dosing (400 mg) of celecoxib daily in patients diagnosed with colorectal polyposis and treated over prolonged periods of three years significantly decreased the recurrence of adenoma and advanced adenoma, when compared to placebo[215]. In follow-up studies, it was reported that although the effects of celecoxib diminished two years after treatment was halted, there was still a considerable treatment benefit at five years[216,217]. Overall, the risk for advanced adenoma relative to placebo was lowered by 52% on low-dose 200 mg bi-daily (vs. 57% at 3 years) and 51% on high-dose 400 mg bi-daily (vs. 66% at 3 years)[217]. Hence, long term use of celecoxib has consistently proven to significantly lower the formation of advanced colorectal cancers in such studies.
We have previously extensively reviewed the outcomes of human clinical trials with celecoxib to treat cancer and which have often shown that sustained, long-term use over several years provided significant benefits in terms of patient outcomes, particularly against metastatic recurrence of cancers[23]. To summarize from the trial outcomes, unfortunately many such clinical trials underway during the last decade suffered from being prematurely terminated because of the growing recognition at the time of cardiotoxic side effects associated with the use of certain COX-2 inhibitors (coxibs), such as Vioxx (rofecoxib)[218]. Although this resulted in a total ban for Vioxx, elevated cardiotoxicity was ruled out for celecoxib in 2015, when it was approved by the USFDA after it was found to be as safe as ibuprofen or naproxen[219]. As an example of one prematurely terminated study, a Phase II trial was reported in 2018 concerning the effects of celecoxib (400 mg twice daily, every day) plus or minus chemotherapy with IFL (irenotecan, 5-fluorouracil (FU), and leucovorin;each cycle comprising IFL over 4 weeks then 2 weeks with no IFL) for previously untreated or unresectable metastatic CRC confirmed by biopsy[155].
Unfortunately, because the Phase II trial was prematurely terminated, only short-term use of celecoxib in patients could be analyzed with follow up to 2 years, and which would not be expected to show significant effects based on the earlier findings outlined above. This Phase II study was further complicated by a protocol variation with the inclusion of 81mg aspirin added with celecoxib to mitigate against possible cardiotoxicity in the high-risk cardiovascular subjects. This trial was limited to a median of three treatment cycles (a total of ~ 20 weeks of daily celecoxib). Nevertheless, the results showed promise in that PFS and overall survival(OS) of patients was improved at 8.7 and 19.7 months, respectively when compared to the historical IFL alone treated controls of 7 and 15 months. After modifying the study protocol, the overall survival was ~91%at one year and 50% at 2 years (median OS = 24.2 months), and the authors could not exclude the possibility of added survival benefit with celecoxib slowing progression of disease. A larger, multicentred Phase III trial(Alliance/SWOG C80702) is underway with FOLFOX (5-FU + oxaliplatin + folinic acid = leucovorin) every 2 weeks for 24 weeks, plus celecoxib (400mg daily treatment extended to 3 years) with longer term follow-up to include 6-year survival and will complete in December 2019.
CONCLUSION
Based on the supportive findings reviewed here, the importance of cytotoxic drugs targeting the CSC population has become self-evident, with significant ramifications for the future of anticancer drug design and treatment. It should be possible to identify and thereby improve the development of drugs used as biological modifiers to eliminate the CSC populations by relying on the selective biomarkers Cripto-1,ABCG2, LGR5 and CD271 to aid in the analysis of CSC drug sensitivity. From the range of evidence provided, it is apparent that such CSC markers are expressed in common across many cancer types and with ESCs. In this manner, the CSC biomarkers can be used to separate enriched CSC populations from the other non-tumorigenic cells and normal stem cells using cell-sorting technology to then identify those drugs with greater potential for specifically and selectively targeting the CSCs by activating their cell death programs.One such drug is celecoxib, with one of its properties being an inhibitor of the COX-2/PGE production by CSCs, otherwise required for promoting greater LGR5 expression and WNT signalling to enhance CSC proliferation.
Cancer treatments have repeatedly failed due to their inability to target and kill the CSCs because these cells are highly resistant to commonly used chemotherapies, having greater survival and metastatic properties.The CSCs can remain dormant, only to then become reactivated, with their capacity for self-renewal and extensive proliferation, metastasis and differentiation giving rise to recurrent tumors, even after extended periods post-treatment. Therefore, it will be essential to be able to identify the CSCs, isolate and study their characteristics in greater detail in order to develop better drug treatments, such as celecoxib, and where the evidence is compelling that such drugs can then either promote or induce the death of the CSCs, both in the circulation and in tumor niches.
DECLARATIONS
Authors’ contributions
Both authors contributed equally to the writing, construction and editing of the manuscript as well as drawing of the figures.
Availability of data and materials
Not applicable.
Financial support and sponsorship
ALHulais RA was supported by the Higher Education of Saudi Arabia (King Abdullah Scholarship) for her PhD program.
Conflicts of interest
Both authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2019.
 Journal of Cancer Metastasis and Treatment2019年1期
Journal of Cancer Metastasis and Treatment2019年1期
- Journal of Cancer Metastasis and Treatment的其它文章
- AUTHOR INSTRUCTIONS
- Call for Papers
- Axillary recurrence after intramammary sentinel lymph nodes metastases with capsular extravasation
- A non-toxic approach for treatment of breast cancer and its metastases: capecitabine enhanced photodynamic therapy in a murine breast tumor model
- A pilot study for distinguishing basal cell carcinoma from normal human skin tissues using visible resonance Raman spectroscopy
- Mass spectrometry-based metaboIomic profiIing of prostate cancer - a piIot study