
运动心肌保护效应:microRNA的调节作用
2019-07-29 01:50陈亚丽王世强李丹王国军
中国运动医学杂志 2019年6期
陈亚丽 王世强 李丹 王国军
1 上海体育学院运动医学康复中心(上海200438)
2 上海上体伤骨科医院(上海200438)
3 湖南工业大学体育学院(株洲412000)
心血管疾病(CVD)的致残率和致死率高、花费大、治疗费用大,已经成为危害人们健康的主要疾病。据《中国心血管病报告2017》数据,我国心血管病患病人数已达2.9亿,严重影响我国人民群众的身体健康。研究心血管疾病的发病发展机制有助于开发有效的心血管疾病的治疗药物,有助于探寻新的心血管疾病的预防手段和策略。
长期规律的有氧运动能够有效降低心血管疾病的发病率,减轻疾病的症状,对心血管疾病具有良好的预防和治疗作用。长久以来,研究人员对运动增强心肌保护效应、促进心脏健康的生物学机制进行了深入研究。近年来,研究发现运动对多个心肌微小RNA(microRNA,miRNA)具有调节作用,可能介导了运动心肌的保护效应,为运动心脏的研究提供了新的视角。心血管疾病发生发展过程中,伴随着病理性心肌重塑。病理性心肌重塑包括心肌肥厚、心肌细胞凋亡、心肌纤维化等环节[1]。研究表明,运动通过miRNA 对心肌细胞肥大、细胞增殖、间质重塑、细胞凋亡、血管再生和电重塑等过程进行调节,产生运动性心脏重塑[2]。近期研究报道,运动改善心肌缺血再灌注损伤、心肌梗死、糖尿病性心肌病、心衰等心脏疾病症状,miRNA可能介导了运动心肌保护作用,进而抑制病理性心肌重塑[3-6]。本文从心肌细胞肥大、心肌细胞增殖、细胞凋亡、心肌血管再生和心肌纤维化等生物过程,综述了miRNA 的运动心肌保护效应。本文应用Pub Med、Medline、web of science、知网数据库检索2008年5月至2018年5月关于运动心肌与miRNA 相关方面的文献,以“exercise,microRNA,cardiac hypertrophy or fibrosis or proliferation or apoptosis or angiogenesis”为英文检索主题词,以“运动,micorRNA,心肌肥大或纤维化、细胞增殖、细胞凋亡、血管新生”为中文检索主题词。通过阅读标题和摘要进行初筛,初步检索得到英文文献122篇,中文10篇,排除与本主题不相关的内容,最终纳入52篇符合标准的文献。
1 microRNA与心脏疾病
miRNA是包含大约22个核苷酸的非编码RNA,从1993年第一个miRNA 被Lee 等发现以来,研究人员发现了38589 条miRNA(截止到2018年5月,http://www.mirbase.org/)。研究发现,miRNA 在心脏发育、心肌细胞分化凋亡、心肌细胞增殖、血管再生、心肌肥大等多个生理和病理过程中具有重要作用。如表1所示。
1.1 miRNA与心肌肥大
病理性心肌肥大是多种心脏疾病常见的病理变化,表现为心肌增厚、心肌扩张和心脏衰竭。近些年发现多个miRNA 在病理性心肌肥大过程中具有调节作用。包括抑制病理性心肌肥大的miRNA,如miR-1、miR-133a 等[7,8];促进心肌细胞肥大的miRNA,如miR-208a、miR-155等[9,10]。
人体和动物模型研究发现,miR-1 通过靶向调节多个信号通路抑制心肌肥大。Ikeda等在新生大鼠心肌细胞中发现,miR-1 通过下调肌细胞增强因子2A(Mef2a)抑制心肌细胞肥大。而在心衰的大鼠模型中,miR-1 呈现低表达,对Mef2a 的抑制作用减弱,进而介导钙调磷酸酶/活化T 细胞因子(CaN/NFAT)信号通路诱导小鼠心肌肥厚[7]。Fibullin-2(Fbln2)是miR-1的另外一个靶基因,在主动脉缩窄诱导的左心室肥大大鼠模型中,miR-1的表达显著降低,通过尾静脉注射结合miR-1的慢病毒后,左心室肥厚程度得到抑制,心肌功能得到改善。进一步发现,miR-1可能通过激活MAPK信号通路防止病理性心肌重塑[8]。Diniz 等离体实验发现,miR-1在甲状腺激素诱导的心肌细胞肥大过程中,呈现低表达状态,miR-1 的过表达抑制了甲状腺素诱导的心肌细胞肥大。在体实验进一步证实,miR-1 通过靶向作用于HADC4 mRNA 起抑制心肌细胞肥大的作用[11]。研究发现,miR-1在主动脉缩窄诱导的病理性心肌肥大的大鼠模型中表达显著下调,心肌细胞横截面积显著增加。离体研究确认了线粒体钙单向转运体(mitochondrial calcium uniporter,MCU)是miR-1 的调节靶基因,结果提示,miR-1的下调通过提升MCU的表达,促进细胞Ca2+的摄取能力,促进心肌细胞肥大。研究同时发现,在运动性心肌细胞肥大模型中,miR-1/MCU通路与主动脉缩窄诱导的病理性心肌肥大具有相同的变化,值得进一步深入研究[12]。
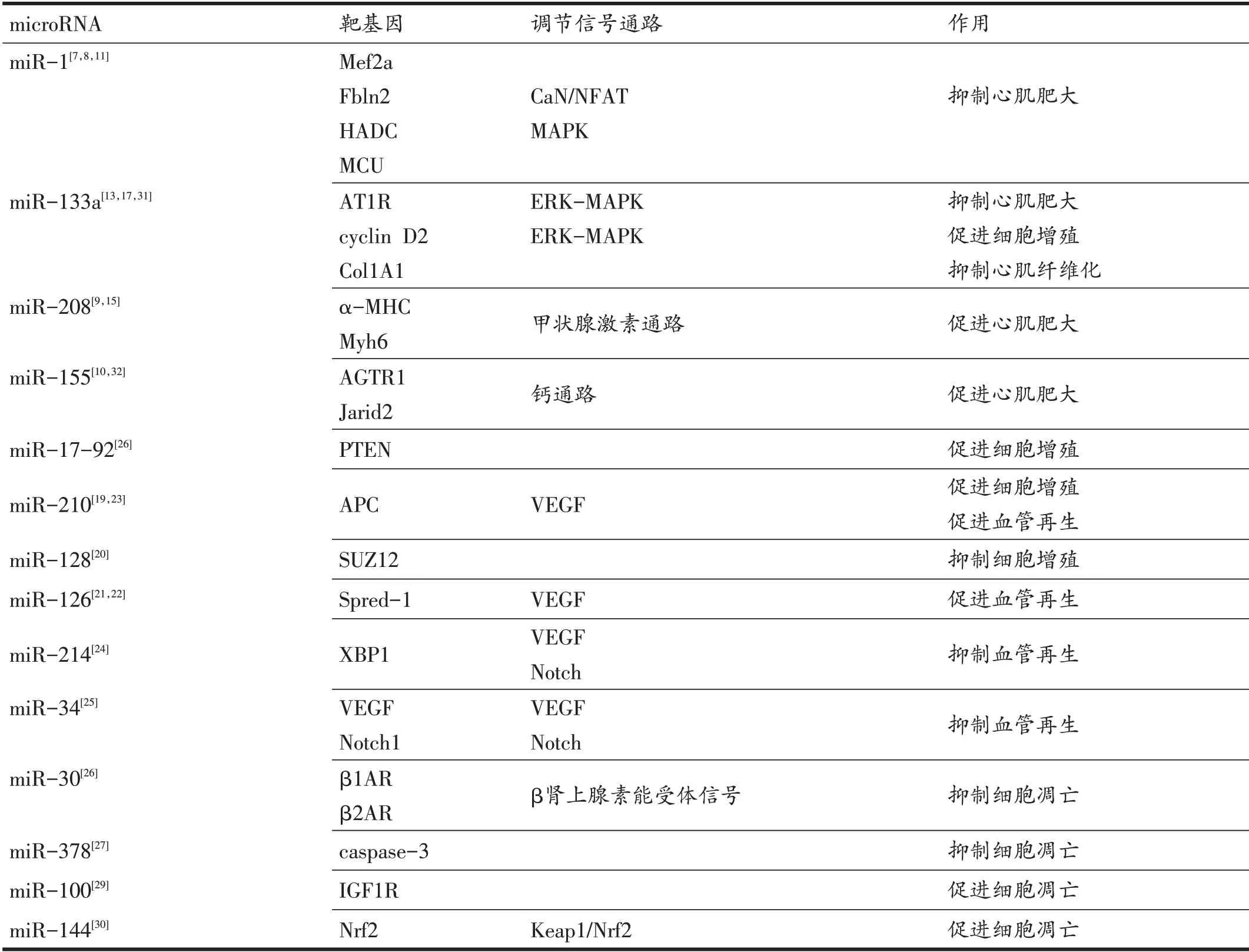
表1 microRNA在病理性心肌重塑过程中的调节作用
与miR-1 类似,miR-133a 也起到抑制心肌细胞肥大的作用。在血管紧张素Ⅱ(Angiotensin Ⅱ,Ang Ⅱ)诱导的心肌细胞肥大模型中,miR-133a 表达显著降低,上调miR-133a可抑制心肌细胞肥大。进一步研究研究表明,miR-133a 可能通过激活MAPK 通路,减少ERK1/2 的磷酸化,抑制心肌肥大程度[13]。与此一致,Diniz 等发现miR-133a 在甲状腺激素诱导的心肌肥大模型中miR-133a的表达显著降低。进一步研究发现,miR-133a 通过靶基因Ⅰ型血管紧张素Ⅱ受体(Type 1 Angiotensin Ⅱreceptor,AT1R)起作用。人体实验发现,血液miR-133a含量与左心室和室间隔心肌肥厚程度成负相关,可以作为预测心肌肥厚的生物标志物[14]。
Diniz 等的另一项研究报道发现,甲状腺功能亢进引起心肌肥大过程中伴随心肌miR-208a的表达上调,靶基因α-MHC 的表达显著下调,而与miR-208b/β-MHC 无关。深入研究发现,α-MHC 通过促进AT1R 表达诱导心肌肥大[9]。Callis 等研究发现,miR-208a 的转基因过表达小鼠则表现为心肌肥厚性增长,并诱发了心律失常,Myh6可能是其作用的靶基因。miR-208a敲除的小鼠则表现为正常的心肌生长和正常的心律[15]。miR-155 也是诱导心肌肥大的miRNA。Yang 等通过Ang Ⅱ诱导H9C2心肌细胞肥大模型,转染miR-155类似物或抑制剂,观察细胞肥大变化情况。结果发现,miR-155 联合Ang Ⅱ比单纯Ang Ⅱ更能促进心肌细胞面积的增加。进一步研究显示,miR-155 可能通过下调靶基因AGTR1 抑制钙信号通路促进心肌肥大[16]。与此一致,Seok等也发现,miR-155也能促进心肌细胞肥大。不同的是,作者研究发现Jarid2是miR-155调控的靶基因,抑制miR-155 的表达能有效降低主动脉缩窄诱导的心肌肥大[10]。
1.2 miRNA与心肌细胞增殖
心肌细胞数量减少是多种心脏疾病共有的结构变化,影响了正常的心脏功能,最终导致心力衰竭的发生。通过一定的手段促进心肌细胞的增殖进而改善心脏功能,是促进心脏恢复的重要策略。越来越多的研究证实,miRNA 对心肌细胞增殖具有调节作用。早年研究表明,在大鼠胚胎发育过程中,miR-133a 可诱导心肌细胞的增殖,miR-133a基因敲除的大鼠心肌细胞增殖出现障碍。进一步研究发现,miR-133a可能通过靶向抑制细胞周期蛋白cyclin D2 促进心肌细胞的增殖[17]。Chen 等发现,miR-17-92 簇是心肌细胞增殖的关键调节因子。在胚胎或新生小鼠心脏中剔除miR-17-92 簇,心肌细胞的增殖受到抑制,心脏发育明显减缓。而miR-17-92 簇的过表达能显著促进胚胎、新生和成年小鼠心肌细胞增殖。研究还发现,miR-17-92簇过表达诱导的心肌细胞增殖对心肌梗死损伤具有保护作用。离体研究进一步证实,肿瘤抑制因子同源性磷酸酶-张力蛋白(phosphatase and tensin homolog,PTEN)是miR-17-92 簇的靶基因,介导了miR-17-92簇的心肌细胞增殖[18]。另外,Arif等发现miR-210介导了心肌细胞增殖。在离体培养的细胞中转染miR-210,大鼠心肌细胞出现明显的单细胞核增殖,并伴随心肌细胞面积减少、多核细胞数量降低、细胞凋亡下降。另外,Bcl-2表达增加,而caspase-3表达下降。软件预测分析结果表明,细胞周期抑制因子泛素连接酶(anaphase promoting complex,APC)是miR-210的靶基因。在心肌梗死小鼠模型中发现,miR-210 转基因小鼠表现出明显的细胞增殖,细胞凋亡下降[19]。
Huang等研究显示,从增殖的新生心肌细胞到分化为终末细胞过程中,miR-128 的表达显著上调。在体研究表明,miR-128 过表达损害了新生小鼠细胞增殖和心肌功能。miR-128通过上调多梳蛋白SUZ12 阻碍细胞周期蛋白抑制剂p27 的表达,进而激活细胞周期蛋白Cyclin E 与CDK2 相结合并激活CDK2,驱动细胞进行细胞分裂。该研究提示,miR-128 可作为促进内源性心肌细胞增殖的重要靶点[20]。
1.3 miRNA与心肌血管再生
心肌血管再生增加了心肌细胞的血液循环,对促进心脏康复具有重要意义。研究证实,多个miRNA 参与了血管内皮细胞增殖、微小血管密度和心肌血管形成和再生的调节[21,22]。Arif 等的研究表明,miR-210 除了能够促进心肌细胞增殖,抑制细胞凋亡以外,还能诱导心肌血管再生。miR-210 通过抑制APC 的表达,间接促进血管内皮生长因子(vascular endothelial growth factor,VEGF)的表达,诱导心肌血管再生[19]。与此一致,Fand 等发现,miR-210 激动剂激活了HGF 的表达,进而促进了血管内皮细胞的增殖。在心梗大鼠模型中miR-210 的过表达增加了心肌微小血管密度,改善了心肌功能[23]。miR-126 促进了血管内皮细胞的增殖,维持了血管的稳定性,在低氧或血管损伤模型中,miR-126 的高表达通过Spred-1 激活了VEGF 血管内皮细胞的增殖信号,促进了血管的形成[22]。人体研究也发现,miR-126 在慢性心衰病人血管形成的早期细胞(angiogenic early outgrowth cells,EOCs)中明显减少。抑制miR-126 损害了EOCs 的血管形成和改善心脏功能的能力[21]。
另外一些microRNA 抑制血管内皮细胞的增殖和血管形成。Duan等研究表明,在心衰小鼠模型中,心肌miR-214 的表达显著下降,腺病毒转染miR-214 的抑制剂抑制了异丙基肾上腺素诱导的心肌血管再生损害,改善了心脏功能。而miR-214 的过表达通过靶向抑制转录因子XBP1的表达,抑制了内皮细胞增殖和血管形成[24]。研究表明,miR-34家族(miR-34a、miR-34b和miR-34c)介导了病理性心肌重塑,通过LNA-antimiR-34 抑制miR-34 家族的表达,增加了VEGF 和Notch1 的表达,激活了VEGF 和Notch 信号通路,促进了心肌血管再生,改善心肌功能,抑制心肌梗死诱导的病理性心脏重塑[25]。
1.4 miRNA与心肌细胞凋亡
Roca-Alonso等证实,miR-30在心肌梗死心脏衰竭动物模型中均显著下调。进一步发现,miR-30可能通过抑制β1肾上腺素能受体(β1- and β2-adrenoceptor,β1AR 和β2AR)阻断β肾上腺素能受体信号,进而抑制心肌细胞凋亡[26]。Fang 等证实,在心肌缺血大鼠模型和1%低氧浓度下的H9C2 细胞模型中,miR-378 的表达显著下调。miR-378 的过表达增加了细胞活力,抑制了细胞凋亡和坏死。而抑制miR-378加重了低氧诱导的细胞凋亡和损伤。生物信息预测和荧光素酶报告进一步证实,细胞凋亡关键调节因子caspase-3 是miR-378的靶基因[27]。另外,有研究发现,miR-21具有显著抑制心肌细胞凋亡的作用。在H9C2细胞模型中,氧化应激诱导了细胞凋亡和miR-21的显著下调,miR-21 过表达抑制了caspase-3 的表达。研究进一步确认程序性细胞死亡基因4(programmed cell death 4,PDCD4)是miR-21的靶基因,miR-21/PDCD4是抵抗氧化应激诱导细胞凋亡的重要通路[28]。
另外一些microRNA 则起抑制细胞凋亡的作用。Chen 等研究证实,miR-100 在H2O2诱导的心肌凋亡过程中显著上调,且具有时间依赖性。抑制miR-100 阻碍了心肌细胞凋亡。进一步研发发现,在低氧诱导的心肌凋亡过程中,miR-100 通过抑制胰岛素样生长因子受体1(insulin-like growth factor 1 receptor,IGF1R)起作用。抑制IGF1R 表达增加了心肌细胞凋亡,去除了miR-100下调诱导的心肌细胞保护效应[29]。Yu等发现,miR-144 拟似物加重了高糖诱导的ROS 生成和心肌细胞凋亡,而miR-144 抑制剂通过激活Nrf2 信号通路,减少ROS 生成,抑制了心肌细胞凋亡,改善STZ 诱导的糖尿病小鼠心肌细胞凋亡[30]。
2 microRNA的运动心肌保护效应
运动能够通过促进心肌细胞增殖、抑制细胞凋亡、增加血管生成、阻碍间质纤维化等环节,产生心肌保护效应,抑制糖尿病、缺血再灌注、高血压、心肌梗死、心脏衰竭等疾病诱导的病理性心肌重塑,改善心脏功能。近年研究证实,miRNA通过降低心肌细胞凋亡、促进心肌细胞增殖、增加心肌血管生成和抑制心肌纤维化介导了运动引起的心肌保护效应[33]。
2.1 miRNA介导运动抗心肌细胞肥大
病理性心肌肥大伴随心肌细胞体积增加、细胞直径和长度增加,即细胞肥大,分子水平上表现为肥大基因ANP/ANF、β-MHC 等被激活而表达上调。研究发现,运动在一定程度上可通过调节miRNA 抑制心肌细胞肥大,降低病理性心肌重塑。研究发现,在肥胖诱导的心脏病理肥大模型中,miR-208a 表达上调,靶基因Med13 含量相应减少,β-MHC 表达增多,持续10 周的运动训练降低了大鼠体重,减少了心肌miR-208a 含量,通过增加Med13 纠正β-MHC 的含量,降低心肌细胞肥大程度,抑制病理性心肌重塑[6]。医学临床和动物研究发现的对病理心肌肥大具有重要调节作用的miR-1 和miR-133a,是否介导了运动抗心肌肥大的过程,目前尚未发现相关的研究报道。
2.2 miRNA介导运动抗心肌细胞凋亡
研究表明,中小强度的有氧运动不仅能降低正常个体心肌细胞的凋亡发生率,而且还能抑制心肌梗死、心肌缺血和糖尿病等诱导的心肌细胞凋亡程度,减轻心肌损伤程度。近年研究发现,miRNA 介导了运动抗心肌细胞凋亡效应。如表2。

表2 运动通过miRNA抑制病理性心脏重塑
Souza等对心脏衰竭大鼠进行有氧运动干预后,通过基因芯片扫描发现,有17 个miRNA 显著上调,14 个miRNA 显著下调。miRWalk 软件预测发现,这些变化的miRNA 主要通过TGF-β1、炎症、凋亡、细胞增殖等途径介导运动对心脏衰竭的保护[3]。Zhao 等发现,8周的游泳运动增加了心肌miR-499 的表达,进而抑制Drp-1的表达,降低了心肌凋亡程度[34]。在另一项研究中发现,运动促进了心肌miR-21 和miR-30b 的表达,其可能通过调节PDCD4和Drp-1抑制小鼠心肌细胞凋亡[35]。在肺动脉狭窄诱导的右心室重塑动物模型中,miR-21表达下调,心肌细胞凋亡增加。运动可以显著上调miR-21,通过抑制PTEN激活PI3K/Akt信号通路,抑制心肌细胞凋亡,改善右心室功能[36]。
中低强度运动可抑制细胞凋亡,而大强度剧烈运动可能诱导心肌细胞凋亡,细胞凋亡对运动强度较为敏感,miRNA 是否参与了该过程的调节,尚不清楚[37]。前期研究曾发现,不同运动强度对microRNA的影响不同,其与细胞凋亡的关系如何,目前尚无相关的研究报道,将是未来研究方向之一[38,39]。
2.3 miRNA介导运动诱导的心肌细胞增殖
研究表明,运动诱导心肌细胞增殖对于运动性心肌肥大不是必要条件,而运动通过诱导心肌细胞增殖产生心肌保护效应,进而抵抗心肌缺血病理性心脏重塑,则不可或缺。多个miRNA参与了该过程的调节。
miR-222 是最受关注的可促进心肌细胞增殖的miRNA之一。Liu等发现,在跑台和游泳诱导的运动性心肌肥大小鼠模型中,miR-222 均呈现显著上调。体外研究发现,miR-222 过表达促进了心肌细胞增殖和体积增加,而抑制miR-222 显著降低了心肌细胞数量和细胞体积。LNA-antimiR-222 在体抑制心肌miR-222的表达,游泳运动诱导的心肌细胞增殖和体积增加明显受到抑制。进一步研究发现,HIPK1 和HMBOX1是miR-222 的靶基因。在心肌缺血模型中,miR-222转基因小鼠表现出较低的细胞凋亡率、较少的心肌梗死面积和心脏功能受损。结果提示,运动诱导的心肌miR-222 上调能够通过促进心肌细胞增殖,降低心肌缺血导致的病理性心脏重塑程度[5]。Shi 等发现miR-17-3p也介导了运动产生的心肌保护效应。研究表明,体内抑制miR-17-3p 的表达,运动诱导的心肌细胞增殖明显受到抑制。尾静脉注射agomir-17-3p增强了心肌细胞增殖,产生心肌保护效应,抑制缺血再灌注诱导的病理性心肌重塑。体外研究表明TIMP-3 可能是miR-17-3p 的调节靶基因,结果提示,运动通过上调miR-17-3p抑制TIMP-3增加心肌细胞增殖,抵抗缺血造成的病理性心脏重塑[40]。
以上两项研究发现,miR-222 和miR-17-3p 通过调节靶基因,进而促进心肌细胞增殖,并通过功能获得或缺失的方法,进一步确认了以上两种microRNA的关键作用,在运动心脏研究领域产生了较大的影响,这也为运动心肌microRNA领域的研究提供了借鉴和参考。
2.4 miRNA介导运动诱导的心肌血管生成
miR-126能间接调节VEGF的表达,是目前发现的介导运动诱导血管生成最重要的miRNA。Ghorbanzadeh等发现,8周的自主转轮运动促进了大鼠心肌miR-126的表达,增加了心肌血管密度[41]。另外一项研究发现运动促进了小鼠心肌miR-126 的表达,靶基因Spred1的表达受到抑制,促进了血管内皮细胞增殖,增加心肌血管密度,降低心肌梗死面积,改善心梗大鼠心脏功能[42]。与此一致,DA 等进一步明确中等负荷运动和大负荷运动使大鼠心肌miR-126 分别上调26%和42%,血管密度分别增加58%和101%,VEGF的表达分别增加42%和108%。其靶基因Spred-1分别下调41%和39%,进而激活了促血管新生信号通路Raf-1/ERK1/2。另一个靶基因PI3KR2 分别下调39%和78%,同时P13K/Akt/eNOS 信号通路相关蛋白显著增加[43]。田振军和宋伟等也发现持续和间歇运动激活具有内皮细胞特异性的miR-126,激活了下游PIK3R2/Spred1 通路,促进心梗大鼠心肌梗死边缘区血管新生,提升心功能,产生心肌保护效应[44]。
2.5 miRNA介导运动抗心肌纤维化
虽然过度运动会产生心肌胶原蛋白过度增多的风险,但中低强度的有氧运动改善心肌间质纤维化程度,是运动心肌保护效应的重要方面。近些年,研究证实运动可通过调节miRNA,降低心肌梗死等心脏疾病诱导的心肌纤维化。Melo 等研究表明,与未经运动的心梗大鼠相比,运动训练使心梗大鼠梗死边缘区域miR-29a和miR-29c的表达分别增加32%和63%,Col1A1和Col3A1分别降低63%和62%,减缓了心肌纤维化程度,改善了心梗大鼠心肌功能[4]。与此一致,肖丽等发现miR-29a 介导了运动抗心肌纤维化作用,miR-101a 也具有重要调节作用。间歇有氧运动抑制了TGF-β1/Smad信号通路,进而上调了心肌miR-29a和miR-101a的表达,降低了心肌Col1A1和Col3A1的表达,抑制了心肌纤维化程度和瘢痕形成,改善了心梗大鼠心脏功能[45]。运动能够抑制糖尿病诱导的心肌纤维化,Chaturvedi 等的研究证实miR-29b 和miR-455 参与了该过程的调节。该研究发现,运动促进了包含miR-29b和miR-455的外泌体的释放,而两者的共同靶基因MMP-9的表达和活性均受到运动干预的抑制,糖尿病性心肌纤维化得到明显改善[46]。
miR-29是调节胶原蛋白的关键microRNA,运动对其家族均有调节作用,介导了运动抗心肌纤维化。miR-21是另一调节心肌纤维化的关键microRNA,其可通过促进心肌成纤维化细胞的增殖增加胶原蛋白的合成。本研究团队前期研究发现,大强度运动通过TGFβ1促进了心肌miR-21的表达,可能是运动性心肌纤维化产生的原因之一[38]。同时,其他学者的研究发现,中强度有氧运动可通过miR-21抑制PDCD4,进而降低心肌细胞凋亡[35]。可见,运动对心肌miR-21具有怎样的调节作用,仍需深入研究。
3 总结与展望
心肌肥大、心肌纤维化、心肌细胞增殖与凋亡、心肌血管新生等生理病理变化在心肌缺血、心肌梗死、糖尿病性心肌病等心脏疾病的发生发展和康复中具有重要作用。miRNA在以上多个环节参与调控了心脏疾病的发生发展,研究发现,miR-1、miR-133a、miR-21、miR-29、miR-214 等miRNA 通过作用于一个或多个靶基因,并通过CaN/NFAT等信号通路参与调节了病理性心脏重塑的发生发展。运动通过下调miR-208a 促进Med13的表达,纠正β-MHC的含量,降低心肌细胞肥大程度。运动通过上调miR-21、miR-222、miR-17-3p、miR-126、miR-29、miR-101a等miRNA抑制细胞凋亡、促进细胞增殖,增加血管新生,减轻心肌纤维化,进而抑制病理性心脏重塑。其中,运动通过miRNA 促进细胞增殖的研究较为深入,通过miRNA 调节心肌细胞纤维化的研究也较为全面,而在血管新生和细胞凋亡方面进行的相关研究较少,也较为浅显,需要进行深入的研究探讨。某些miRNA,如miR-21可能对运动强度较为敏感,深入研究不同运动强度对这些miRNA 的影响及作用,将有助于进一步丰富和发展运动心肌保护效应的生理学机制。
尽管不少研究发现miRNA 介导了运动心肌保护效应,但miRNA 心肌保护效应的调节机制尚不清楚,多数研究停留在表达变化的层面,缺乏通过miRNA 的“功能获得或缺失(gain or loss of function)”对其调节机制进行深入的研究。近些年研究发现的外泌体囊泡包含并可分泌microRNA,外泌体来源的microRNA 对病理心肌重塑的调节,以及在运动心肌保护效应中的作用将是未来的研究热点。另外,目前的研究多是在动物研究上进行的,尚缺乏运动对miRNA 的调控与人体心脏健康的关系研究,哪些miRNA 可以作为运动干预心脏疾病效果评价的生物标志物,也将成为未来的研究热点。进一步探究miRNA 在心脏疾病发生过程中的调节机制,探寻准确的治疗靶点和干预手段抑制心肌细胞肥大,减少细胞凋亡,减轻心肌纤维化,增加心肌细胞增殖,促进心肌血管再生,对于控制和减缓病理性心脏重塑进程、促进心脏康复具有重要意义。
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
中国慈善家(2021年5期)2021-11-19
感染、炎症、修复(2021年1期)2021-07-28
昆明医科大学学报(2021年2期)2021-03-29
昆明医科大学学报(2021年2期)2021-03-29
心肺血管病杂志(2020年5期)2021-01-14
妇女(2020年4期)2020-04-20
科学咨询(2020年10期)2020-04-01
英语文摘(2019年6期)2019-09-18
体育科学(2018年12期)2019-01-04
