
2019年4月美国和欧盟新批准药物概述
2019-07-15 07:53孙友松
药学进展 2019年5期
孙友松
(上海递鸽医药科技有限公司,上海 201210)
1 2019年4月美国FDA新批准药物
2019年4月,美国FDA共批准13个新药,包括1个全球首次批准的新分子实体[厄达替尼(erda fitinib,1)]、1个全球首次批准的新生物制品(intravenous immune globulin)、2个美国首次批准的新生物制品(romosozumab-aqqg、risankizumab-rzaa)、1个 美 国撤市后重新上市的药品(替加色罗)、2个新复方、1个新生物类似药和5个新增适应证药物(见表1)。
1.1 Intravenous immune globulin
Intravenous immune globulin是一种静脉注射用免疫球蛋白,由ADMA生物公司研发,研发代号为RI-002,商品名为Asceniv®。2019年4月1日,美国FDA批准Asceniv用于治疗成年人或12 ~ 17岁青少年患者的原发性体液免疫缺陷病,预计该产品将于2019年下半年上市销售[1]。
Asceniv此次获得美国FDA批准基于1项开放性、多中心关键Ⅲ期临床研究(NCT01814800)结果。该研究招募了美国9个中心超过59例原发性体液免疫缺陷病受试者,受试者在一年内定期输注Asceniv,并评估受试者的严重细菌感染率。研究结果显示,59例原发性体液免疫缺陷病受试者均未发生严重细菌感染,达到临床研究的主要终点;临床研究的次要终点包括由于感染而无法工作的天数、医生的不定期访问天数及因感染而住院的天数,与其他治疗药物发表的临床研究结果相比Asceniv具有明显优势[2]。
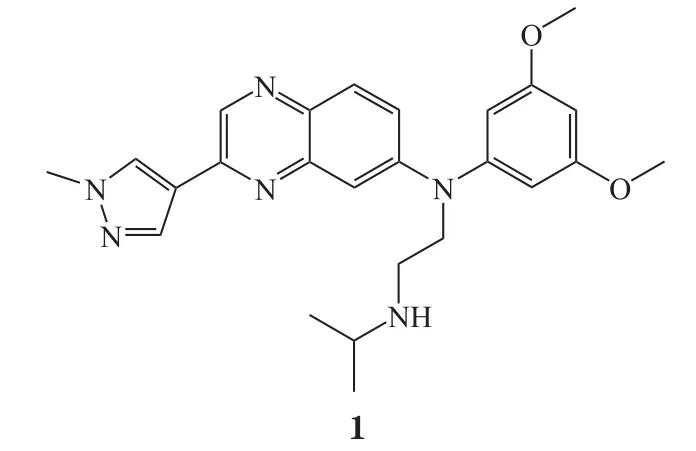
1.2 厄达替尼
厄达替尼是一种FGFR激酶抑制剂,由杨森公司研发,商品名为Balversa®。2019年4月12日,美国FDA批准厄达替尼用于二线治疗铂类化疗失效的局部晚期或转移性尿路上皮癌,患者携带FGFR3或FGFR2基因突变[3]。厄达替尼是美国FDA批准的首个FGFR激酶抑制剂,也是美国FDA批准的首款针对转移性尿路上皮癌的靶向疗法。2018年3月,美国FDA授予厄达替尼突破性疗法资格认定(breakthrough therapy designation,BTD);2018年9月,美国FDA授予厄达替尼优先审评资格(priority review designation)。美国FDA同时批准了一款凯杰(Qiagen)公司的伴随诊断试剂Therascreen®,该伴随诊断试剂基于PCR(polymerase chain reaction, 多聚酶链式反应)技术,用于诊断患者的FGFR基因突变情况。
厄达替尼此次获得美国FDA批准基于一项多中心、开放性、单臂Ⅱ期临床研究(BLC2001研究,NCT02365597)结果,该研究招募了87例局部晚期或转移性尿路上皮癌受试者。临床研究结果显示,厄达替尼治疗组总缓解率(objective response rate,ORR)为32.2%,其中2.3%的受试者达到完全缓解,且厄达替尼对PD-1/PD-L1疗法失效的受试者仍有应答;此外,受试者中位缓解持续时间(duration of response,DoR)长达5.4个月[4]。
2 2019年4月欧盟新批准药物
2019年4月,欧盟委员会(European Commission,EC)共批准9个新药,包括1个全球首次批准的新分子实体[索格列净(sotagli fl ozin,2)]、2个欧盟首次批准的新分子实体(达克替尼、扎那米韦)、2个欧盟首次批准的新生物制品(risankizumab、andexanet alfa)、1个新复方、2个新生物类似药和1个新增适应证药物(见表2)。
索格列净是一种SGLT1/SGLT2双靶点抑制剂,由赛诺菲公司和Lexicon公司共同研发,研发代号为LX4211,商品名为Zynquista®。2019年4月26日,欧盟委员会批准索格列净用于治疗1型糖尿病。SGLT1靶点影响胃肠道中的葡萄糖吸收,SGLT2靶点影响肾脏的葡萄糖重吸收[5]。
索格列净此次获得欧盟委员会批准基于3项随机、多中心、双盲、安慰剂对照Ⅲ期临床研究(inTandem1研究,NCT02384941;inTandem2研究,NCT02421510;inTandem3研究,NCT02531035)结果,3项研究共招募约3 000例单独使用胰岛素控制血糖效果不佳的成人1型糖尿病受试者,评价索格列净的安全性和有效性。临床研究结果显示,索格列净达到了从基线到24周糖化血红蛋白(HbA1c)含量持续稳定变化的临床研究主要终点。索格列净目前正在开展11项关于2型糖尿病的临床研究[6]。
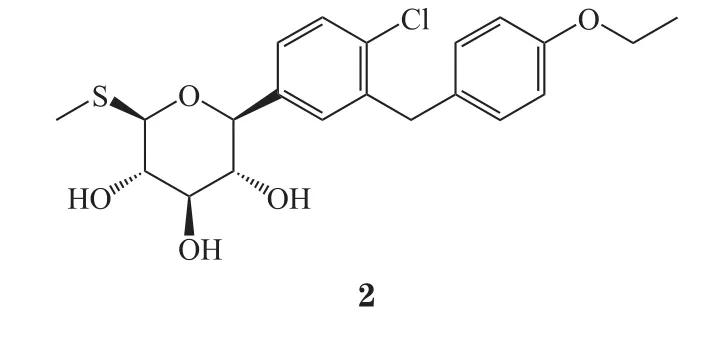
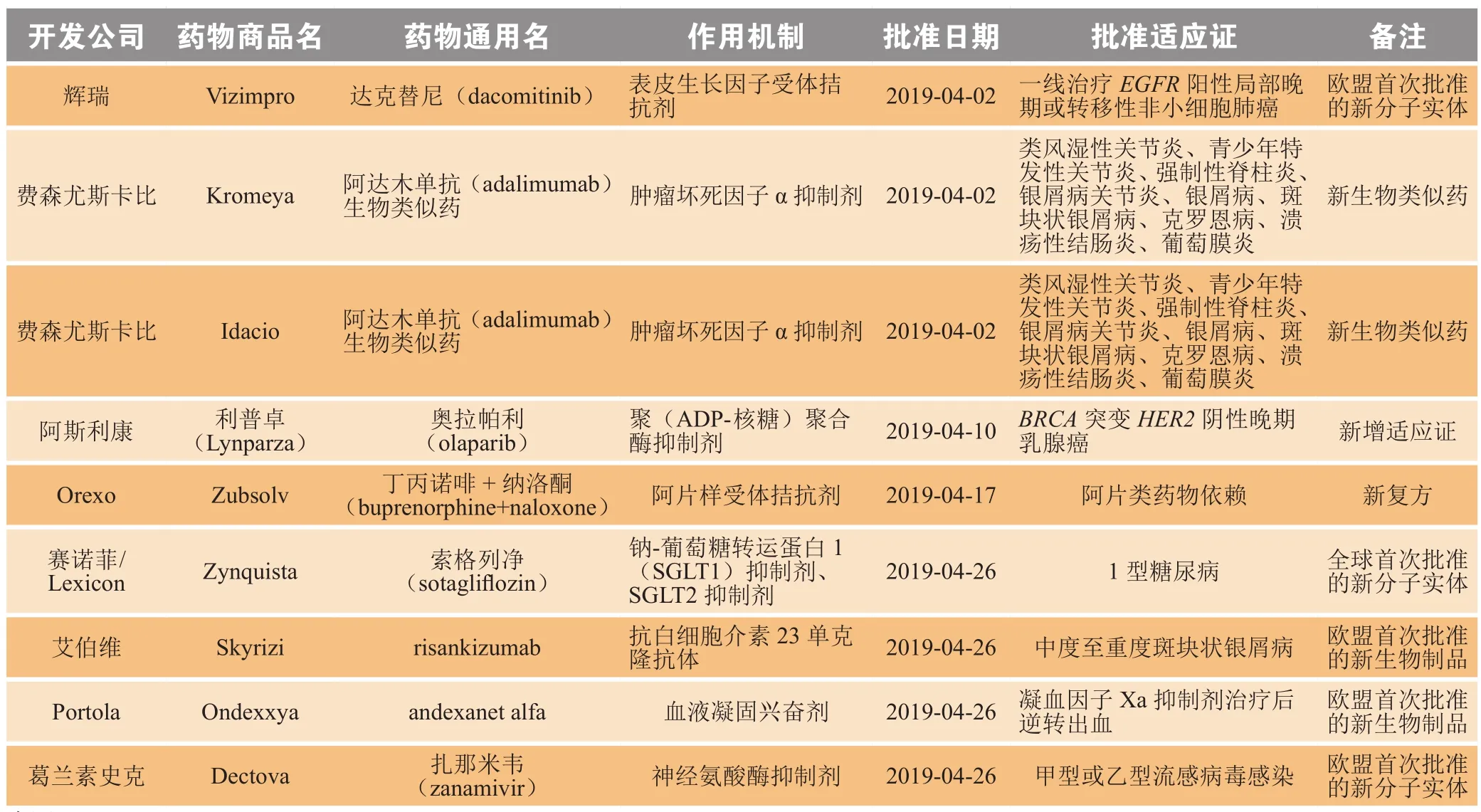
表 2 2019年4月欧盟新批准药物Table 2 New drugs approved by European Commission in April 2019
猜你喜欢
医学概论(2022年3期)2022-04-24
现代临床医学(2022年2期)2022-04-19
中老年保健(2021年3期)2021-12-03
中国生殖健康(2020年7期)2020-12-10
灾害医学与救援(电子版)(2018年1期)2018-06-05
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
医学研究杂志(2015年7期)2015-06-22
中国医学科学院学报(2015年5期)2015-03-01
同位素(2014年2期)2014-04-16
中国中西医结合外科杂志(2013年3期)2013-03-11
