
液态聚碳硅烷的固化及陶瓷化
2019-07-08 02:40:50朱世步孟祥利闫联生张晓虎
航天制造技术 2019年3期
朱世步 张 强 孟祥利 杨 星 闫联生 张晓虎 崔 红
液态聚碳硅烷的固化及陶瓷化
朱世步 张 强 孟祥利 杨 星 闫联生 张晓虎 崔 红
(西安航天复合材料研究所,西安 710025)
采用非等温DSC、TG等研究了SiC陶瓷先驱体-液态聚碳硅烷(LPCS)的固化、陶瓷化行为,运用FTIR、XRD、SEM等手段表征了LPCS先驱体在不同温度的裂解产物结构和微观形貌。通过Kissinger、Ozawa及Crane方程得到LPCS先驱体的固化动力学参数:活化能a=96. 84kJ/mol,反应级数=0.94。LPCS先驱体的失重反应主要发生在400~800℃阶段,先驱体中有机官能团逐渐减少,基本完成无机化转变。XRD结果表明,在1000℃以下裂解得到的产物为表面致密的非晶态SiC结构,而在1500℃下裂解产物发生了晶化转变,得到的陶瓷产物主要为β-SiC相。LPCS具有较低的室温粘度(0.25Pa·s)、良好的室温稳定性,适于用作PIP工艺制备SiC基复合材料的先驱体。
LPCS先驱体;固化动力学;陶瓷化;SiC陶瓷
1 引言
碳化硅(SiC)陶瓷具有高耐磨性、优异的热化学稳定性、抗氧化和抗腐蚀等性能,作为重点材料受到世界范围内的广泛关注[1,2]。其中,连续纤维增强SiC基复合材料具有高比强、低密度、优异的断裂韧性等优势[3],在高温热结构部件具有广泛的应用,如燃烧室、喷管、前缘、鼻锥等[4,5]。目前,制备连续纤维增强SiC陶瓷基复合材料的工艺主要包括化学气相渗透法(chemical vapor infiltration, CVI)[6]、液相渗硅(liquid silicon infiltration, LSI)[7]、聚合物浸渍裂解(polymer impregnation and pyrolysis, PIP)[8]等。CVI工艺能够实现构件的净尺寸成型,但是工艺周期长、成本高昂;LSI工艺通过Si与C的原位反应形成SiC基体,但是高温反应容易造成纤维损伤,同时基体中残余硅难以控制。相比CVI和LSI工艺,PIP工艺具有微观结构可控、成本较低、复杂结构大尺寸成型等优势;同时具备制备温度相对较低,可减少高温对纤维的损伤,能够提升复合材料的综合性能,是制备SiC陶瓷基复合材料迅速发展的方法。
自日本科学家Yajima合成了主链为Si-C结构的聚碳硅烷(polycarbosilane, PCS)以来[9],PCS作为SiC陶瓷先驱体受到了广泛关注[10~12],但该先驱体室温下为固态,作为陶瓷基复合材料浸渍剂使用时存在浸渍效率偏低等不足。SiC陶瓷先驱体应该具有以下特征:良好的工艺性,能制备连续纤维、薄膜及陶瓷基复合材料,陶瓷产率高等。在众多先驱体中,液态聚碳硅烷具有低粘度、大量的活性官能团及接近化学计量比的C/Si比等优点,被认为是最具潜力的SiC陶瓷先驱体。美国聚合物转化陶瓷领域的领导者——Starfire公司成功制备的商业化液态超支化聚碳硅烷陶瓷先驱体(称为SMP)应用于SiC基复合材料的制备[13]。研究表明,液态超支化聚碳硅烷在固化过程中发生氢化加成反应,可显著提升先驱体的分子量同时降低先驱体碎片的气化,其陶瓷产率可超过70%[14,15]。Luo等采用液态SiC陶瓷先驱体(LPVCS)制备了SiC/SiC复合材料,结果表明复合材料的致密效率和密度显著提高[16]。液态聚碳硅烷陶瓷先驱体合成工艺多样、原料广泛,这些因素影响先驱体的分子量、分子结构等。聚合物先驱体的结构不仅影响聚合物转化陶瓷的制备工艺,同时还影响着最终陶瓷产物的组成、相结构、微观结构及性能。因此,开展液态聚碳硅烷先驱体的固化及陶瓷化行为分析具有重要意义。
本文以含有Si-H和-C=CH2基团的新型液态聚碳硅烷先驱体(LPCS)为研究对象,开展了LPCS的固化行为、陶瓷化过程及结构转变研究,表征了其固化前后结构及陶瓷化产物结构,分析了其工艺性能及陶瓷产率,为LPCS先驱体采用PIP工艺制备SiC陶瓷基复合材料提供理论参考。
2 实验
2.1 主要原材料及实验
液态聚碳硅烷(LPCS)的交联固化过程在N2气氛保护下以一定程序升温至300℃保温2h,交联固化得到固化产物。交联固化产物于N2氛围保护下在管式炉中升温至不同的裂解温度(700℃、1000℃及1500℃)热解,热解后得到黑色坚硬产物。
2.2 实验表征
采用TG209F3型(NETZSCH,德国)DSC热分析仪,称量LPCS先驱体样品置于坩埚中,分别以5K/min、10K/min、15K/min及20K/min的升温速率在N2氛围内对其进行升温固化扫描,记录固化反应的起始温度、峰值温度及终止温度;TG/TGA热失重分析,N2气保护,升温速率为10K/min,升温区间为室温至980℃。红外光谱分析(FT-IR)采用PerkinElimer 2000型红外光谱仪表征LPCS先驱体及不同温度处理样品的结构,波数范围为400~4000cm-1。通过德国Bruker公司生产的D8 Advance型XRD衍射仪表征不同裂解温度下裂解产物的晶相结构,2扫描范围为20°~90°。采用Anton Paar公司生产的MCR302型流变仪测试LPCS先驱体的粘温曲线,空气温度条件下以5K/min的升温速率由室温升高至180℃,记录不同温度下先驱体的粘度变化。采用JEOL JSM-64690LV(JEOL)型扫描电子显微镜(SEM)观察碳化产物的微观形貌。
3 结果与讨论
3.1 LPCS的固化行为
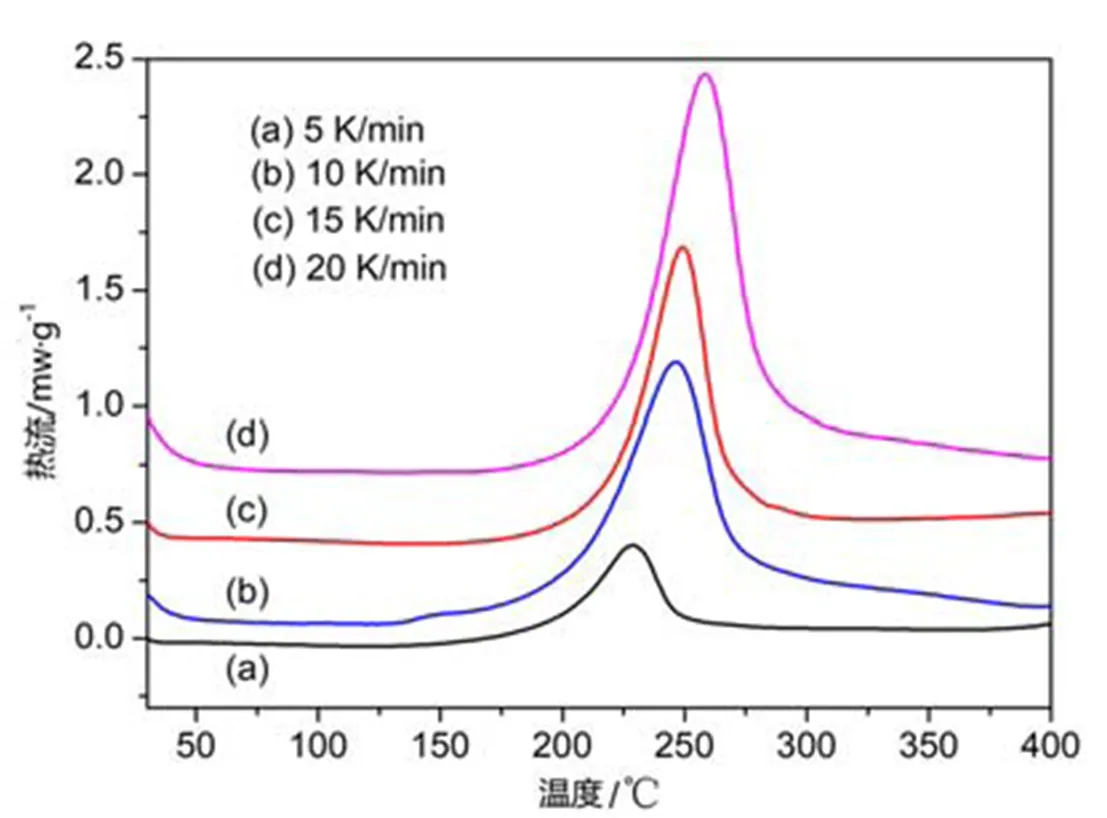
图1 不同升温速率下LPCS先驱体的DSC曲线
LPCS在室温下为流动性良好的液态,可望作为PIP工艺制备SiC基复合材料的先驱体。通过DSC非等温法可以探究LPCS的固化特性,图1表示在不同升温速率下LPCS的DSC曲线。可以发现,LPCS先驱体的固化为防热反应,反应峰值温度出现在220~255℃区间内,随着固化升温速率的提高,放热峰越来越显著,同时峰值温度逐渐向高温方向移动,这是因为随着升温速率的升高,固化反应时单位时间内释放的热量增大(即d/d变大),而LPCS先驱体自身具有温度变化的滞后性,产生更大的热惯性导致固化反应放热峰向着温度更高的方向平移。
对LPCS先驱体进行固化动力学分析能为初步确定先驱体的固化工艺参数提供理论指导。通过Kissinger方程、Ozawa方程和Crane方程可以得到固化反应的表观活化能、反应级数等动力学参数,其中表观活化能a决定反应难易程度、反应级数可推测反应机理,这些参数对于了解LPCS先驱体固化反应有着十分重要的意义。
Kissinger方程[17]:
式中:为升温速率,K/min;p为DSC曲线峰值温度,K;为指前因子;为气体常数,一般取8.314 J/(mol·K);a为固化反应表观活化能,J/mol。依据Kissinger方程,通过ln(/p2)对1/p作图,如图2a所示。采用线性拟合的方法可获得拟合直线,通过所得直线的斜率、截距可算出观活化能(a=94.61kJ/mol)和指前因子(=1.59×109)。
在计算动力学参数时,Ozawa方程避免了对反应机理函数的假设而直接求解反应活化能a值,被广泛应用于验证其它各种假设反应机理函数求出的a值,其公式如下[18]:
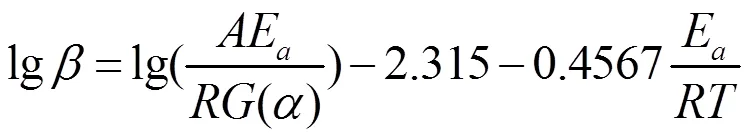
式中,α为固化度;G(α)为积分形式机理函数。根据Ozawa方程,以logβ对Tp作图,可得到近似线性关系,反应活化能Ea可通过拟合直线斜率计算得到Ea为99.07 kJ/mol,与前文采用Kissinger方程计算的活化能Ea值相近。
LPCS先驱体固化反应是过程复杂,涉及众多分子键合形成稳定网状结构的过程,其反应级数的计算则需要借助Crane方程[19],如下所示:
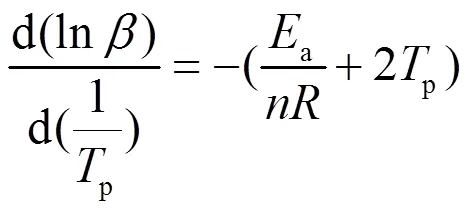
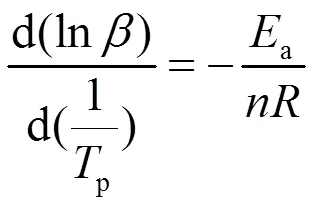
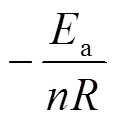
一般采用级固化反应模型描述树脂的固化反应动力学:
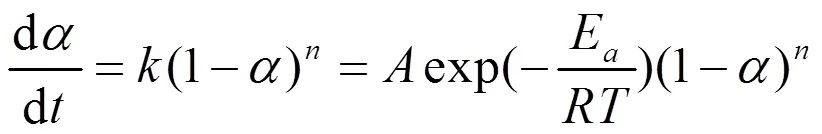
将Kissinger、Ozawa和Crane方程求出的化学动力学参数(指前因子为1.59×109,a取Kissinger和Ozawa方程平均值为96.84kJ/mol,反应级数为0.94)代入式(6),可得LPCS先驱体固化动力学方程:
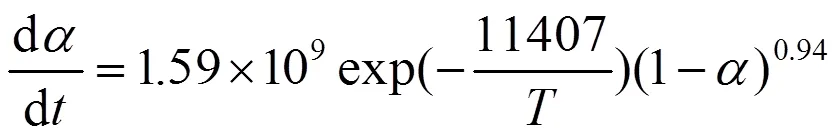
固化度与反应温度及反应时间都有着紧密的关系,反应温度越高,反应时间越长,固化度越大。
LPCS经过交联固化后形成淡黄色固体,通过FTIR表征了固化前后先驱体的结构变化,如图3所示。可以发现,在LPCS先驱体的红外图谱中,3078、1630cm-1分别为-CH=CH2中C-H的伸缩振动峰及C=C双键的伸缩振动峰;2920、2882cm-1出现的吸收峰分别表示的是Si-CH3和Si-CH2-Si中的C-H伸缩振动峰;2131cm-1的吸收峰为Si-H伸缩振动峰;Si-CH2-Si的变形振动峰位于1356cm-1处,1250cm-1特征吸收峰为Si-CH3中C-H的变形振动峰,Si-O-Si键的伸缩振动峰出现在1047cm-1处。由FTIR结果可知,LPCS先驱体分子链中含有Si-CH2-Si,Si-H、Si-CH3等结构单元。对比固化后的LPCS红外图谱,在3440 cm-1和1610cm-1出现新的吸附峰,这可能是制样过程中引入的吸附水中O-H的伸缩振动和弯曲振动造成的[20]。3070cm-1和1630cm-1的-C=CH2中C-H伸缩振动和C=C伸缩振动吸收峰消失,位于2131cm-1的Si-H伸缩振动峰强度明显下降,同时Si-H伸缩振动峰与1250cm-1处Si-CH3变形振动的吸光度比值显著降低,可推测LPCS的固化反应包括Si-H之间的脱氢耦合以及-C=CH2和Si-H键之间的加成反应,先驱体形成空间网络结构,先驱体由液态变成淡黄色不溶不熔固体。

图3 LPCS先驱体及不同温度裂解产物的FTIR图谱
3.2 LPCS陶瓷化行为分析
为了研究LPCS先驱体由聚合物向陶瓷转化的陶瓷化过程,对固化前后先驱体开展了从室温至980℃的TG分析,实验在N2氛围中进行测试,结果如图4所示。由固化前LPCS的TG曲线可知,先驱体在室温至150℃之间微小失重,这部分主要是发生了先驱体小分子挥发等;200~400℃阶段具有大量失重,先驱体的交联固化主要发生在该阶段,先驱体发生脱氢耦合以及-C=CH2和Si-H键之间的加成反应等;400~800℃阶段主要是先驱体的有机官能团的断裂和热分解,产生H2、CH4等小分子气体逸出;高于800℃以后,先驱体从有机物向无机物的转变基本完成,产物为均一的无机物,该阶段先驱体的失重较小,基本处于稳定状态。经过交联固化后的LPCS先驱体,室温至400℃阶段失重较小,这是由于先驱体已经过300℃的交联固化反应,该阶段保持稳定;400℃以后的阶段与固化前LPCS先驱体样品曲线趋势一致,同样发生了先驱体分子侧链有机官能团断裂、有机-无机转化等陶瓷化过程,但是最终陶瓷产率较固化前样品有显著提高,其980℃条件下裂解陶瓷产率可达82.1%。可知,陶瓷先驱体的交联固化过程对陶瓷化过程有着重要意义。
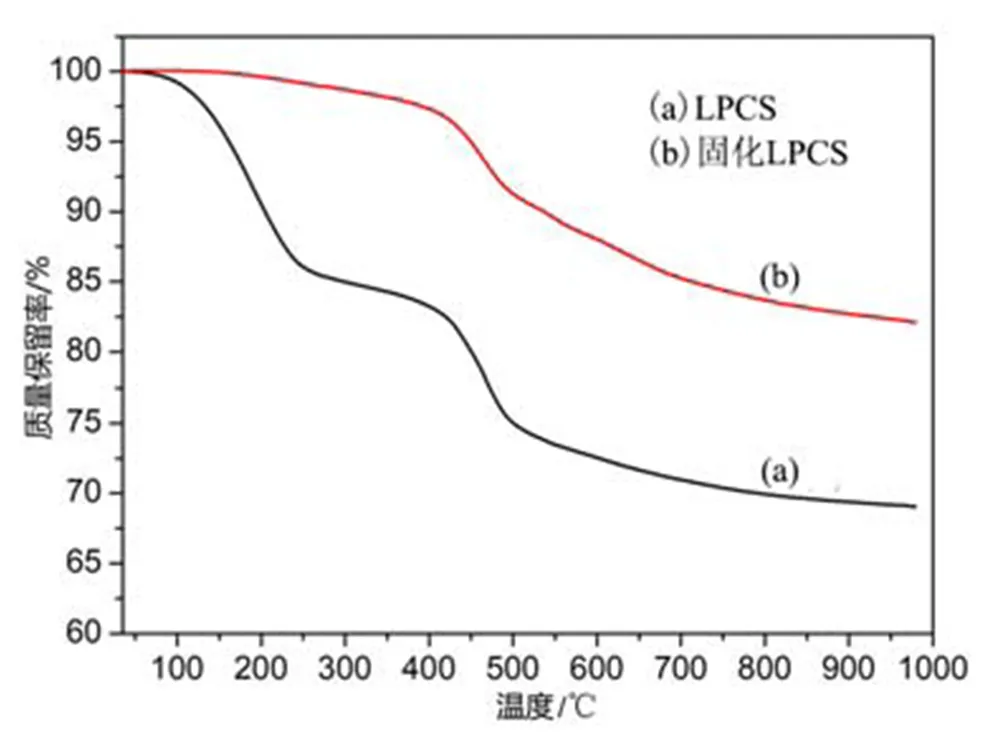
图4 LPCS先驱体交联固化前后TGA曲线
对比研究了不同温度下裂解产物的FTIR图谱,如图3中的(c)、(d)、(e)所示。在700℃条件下裂解产物,先驱体中Si-H等特征吸收峰消失,只在2920cm-1(Si-CH3中的C-H伸缩振动峰)、2882cm-1(Si-CH2-Si中的C-H伸缩振动峰)及1384cm-1(Si-CH3中的C-H弯曲振动峰)出现微弱的吸收峰,同时新出现了983cm-1和778 cm-1附近新的宽峰,分别对应于Si-O-Si和Si-C特征吸收峰,表明在700℃条件下先驱体发生了有机无机转化,有机官能团基本分解,形成无定形结构的SiC,这与前文TG分析结果保持一致;当裂解温度升高至1000℃后,有机官能团完全消失,只出现1087cm-1和820cm-1特征吸收峰,同时与700℃图谱相比,吸收峰半峰宽变窄和出现蓝移现象,表明先驱体在该温度条件下完成了无机化转变;裂解温度进一步提高至1500℃,吸收峰变得更加尖锐、峰宽变窄,先驱体发生了深度陶瓷化转变。
通过XRD对LPCS先驱体在不同热解温度下裂解产物的相结构进行了分析,如图5所示。从图中可以发现,700℃条件下裂解产物为无定形状态,没有出现明显的衍射峰,只在36°附近出现强度很小的宽峰,表明在该温度下裂解形成的是无定形态SiC。1000℃下裂解产物衍射图谱开始出现了位于2=36.5°、60.1°和71.9°的宽峰,分别归属于β-SiC的(111)面、(220)面和(311)面(2=71.9°);同时还出现了石墨(002)晶面(2=26.5°)的衍射峰,说明在该条件下裂解先驱体完成了无机化转化,得到了结晶程度较低的SiC和少部分石墨碳。当裂解温度进一步提高至1500℃,裂解产物位于36.5°、60.1°、71.9°的特征衍射峰强度变强、峰型趋向尖锐,说明1500℃下非晶态陶瓷完成β-SiC微晶形成的晶化过程,陶瓷产物主要为晶化β-SiC。由此可知,先驱体的热解温度对产物的结晶行为具有重要影响。
通过SEM表征了LPCS先驱体不同温度裂解产物的微观形貌,如图6所示。可以发现,在1000℃和1500℃条件下裂解得到陶瓷产物表面致密,没有发现明显的微孔,表明LPCS先驱体能高温裂解形成无孔致密陶瓷,同时基于该先驱体具有高的陶瓷产率,LPCS先驱体有望成为适宜的SiC陶瓷基复合材料的候选先驱体之一。
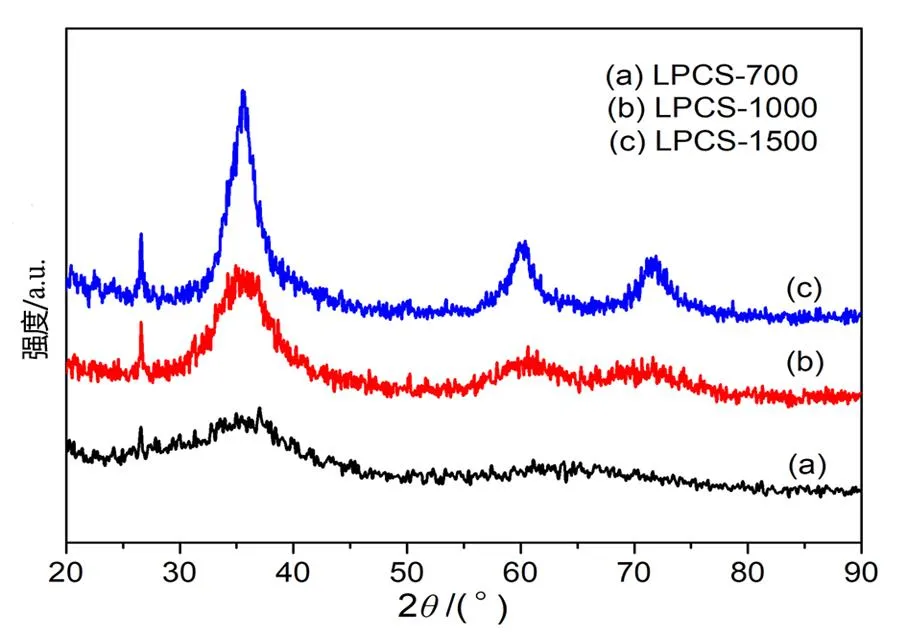
图5 交联固化PLCS先驱体不同温度裂解产物的XRD图谱
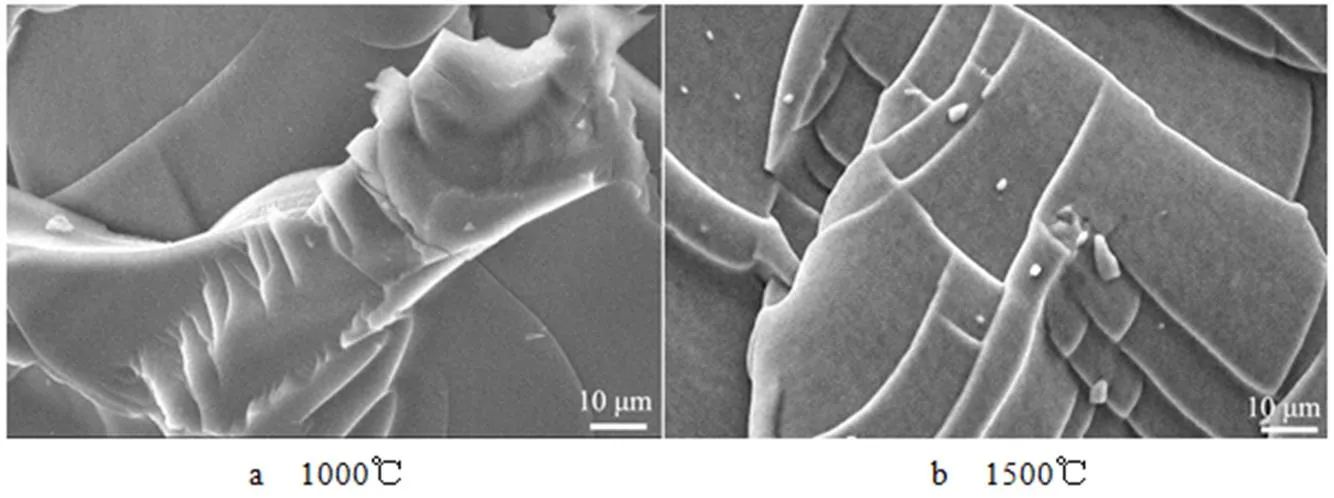
图6 交联固化LPCS先驱体在不同温度裂解陶瓷产物SEM图
3.3 LPCS工艺适用性
首先,对比了水珠与LPCS先驱体在碳纤维织物上的浸润性,如图7所示。由图可知,小水珠与碳纤维浸润性差,液滴与碳纤维表面接触角较大,而LPCS先驱体能够完全铺展在碳纤维织物上,说明其与碳纤维有良好的浸润性,有利于提升PIP的浸渍效率。

图7 水珠和LPCS先驱体液滴滴在碳纤维织物表面的形态
LPCS的室温粘度为0.25Pa·s,粘度低于0.3Pa·s,满足浸渍要求[21];将先驱体在室温下放置45d后,其粘度为0.28Pa·s,同样表现出良好的流变性能,表明LPCS先驱体粘度特性随着存放时间变化不大,便于工程化生产PIP工艺对先驱体的使用与存在。由LPCS先驱体的粘温曲线(5K/min)可知(图8a),随着温度的升高先驱体粘度略有下降,当温度高于160℃后,粘度开始增加,先驱体开始发生固化反应,这与DSC测试结果一致。图8b给出的是LPCS先驱体室温放置前后的FTIR图谱。先驱体在室温放置45d后,其红外特征吸收峰保持一致,说明其结构没有明显变化,进一步说明了LPCS先驱体具有良好的存放性,便于PIP工艺的实施。综合来看,LPCS先驱体具有良好的纤维浸润性、适宜的室温粘度和优异的室温存在稳定性,可作为PIP工艺制备连续纤维增强碳化硅陶瓷基复合材料的候选液态先驱体。
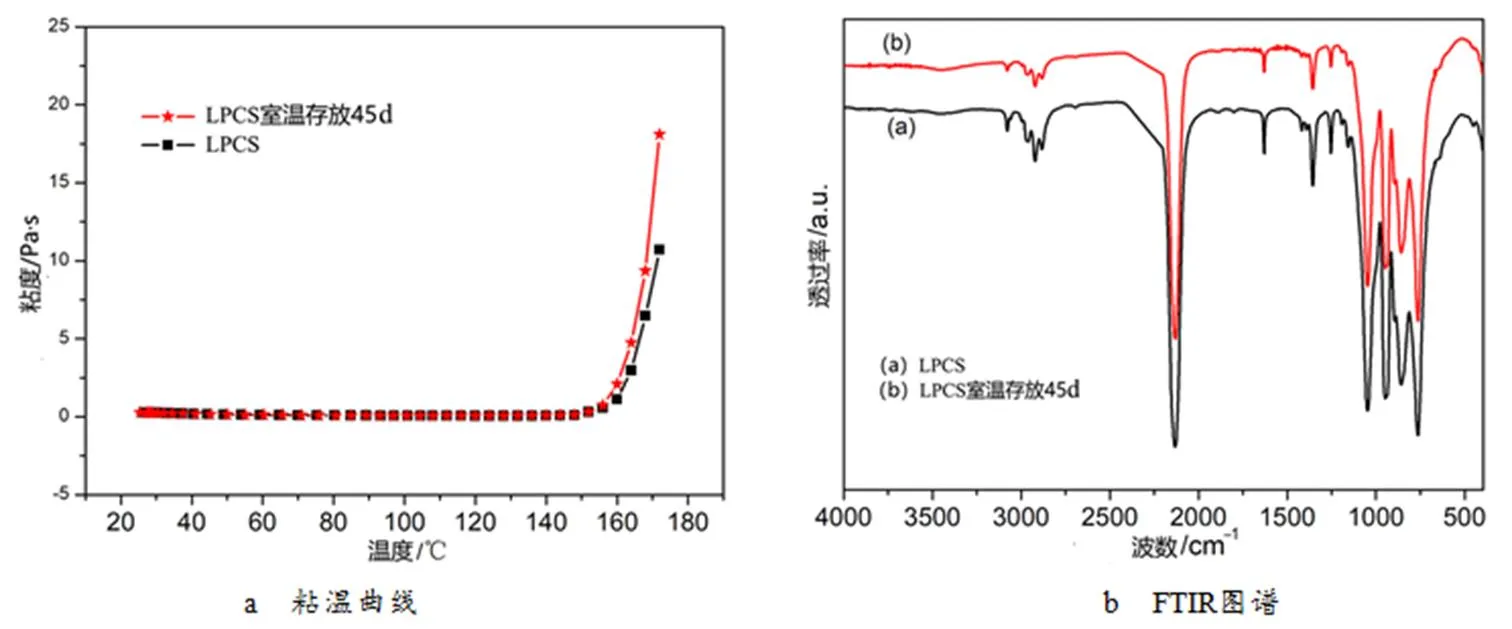
图8 LPCS先驱体室温放置前后的粘温曲线和FTIR图谱
4 结束语
通过DSC、FT-IR、TG、XRD等表征手段对LPCS先驱体的固化、陶瓷化及产物进行了研究,得出以下结论:

b. LPCS先驱体经交联固化后,在不同温度下裂解产物为致密结构,在980℃下的陶瓷产率可达82.1%,1000℃裂解产物为非晶态结构,当温度升高至1500℃后,裂解产物发生晶化转变,产物主要形成β-SiC结构。
c. LPCS先驱体具有良好的纤维浸润性、适宜的室温粘度和优异的室温存在稳定性,可作为PIP工艺制备连续纤维增强碳化硅陶瓷基复合材料的候选液态先驱体。
1 Kim Y W, Lim K Y, Seo W S. Microstructure and thermal conductivity of silicon carbide with yttria and Scandia [J]. Journal of the American Ceramic Society, 2014, 97(3): 923~928
2 Lee L, Kazimi M S. A structural model for multi-layered ceramic cylinders and its application to silicon carbide cladding of light water reactor fuel [J]. Journal of Nuclear Materials, 2015, 458: 87~105
3 Naslain R R. SiC-matrix composites: nonbrittle ceramics for thermostructural application [J]. International Journal of Appllied Ceramic. Technology, 2005, 2: 75~84
4 Lacombe A, Pichon T, Lacoste M. High temperature composite nozzle extensions, a mature and efficient technology to improve upper stage Liquid Rocket Engine performance [C]. AIAA 2007, 2007~5470
5 Naslain R. Design, preparation and properties of non-oxide CMCs for application in engines and nuclear reactors: an overview [J]. Composites Science and Technology, 2004, 64: 155~170
6 Yu Shengjie, Chen Zhaofeng, Wang Yang, et al. Effect of fabric structure on the perneability and regeneration ability of porous SiCf/SiC composite prepared by CVI [J]. Ceramics International, 2019, 45(9): 11564~11570
7 Choi J H, Kim S, Kim S, et al. Thermal and mechanical properties of C/SiC composites fabricated by liquid silicon infiltration with nitric acid surface-treated carbon fibers [J]. Journal of Ceramic Processing Research, 2019, 20(1): 48~53
8 Li Mingyuan, Yang Dexuan. Wang Honglei, et al. Fabrication of a 3D4d braided Sicf/SiC composite via PIP process assissted with an EPD method [J]. Ceramics International, 2019, 45(9): 11668~11676
9 Yajima S, Okamura K, Hayashi J, et al. Synthesis of continuous SiC fibers with high tensile strength [J]. Journal of the American Ceramic Society, 1976, 59(7-8): 324~327
10 Colombo P, Mera G, Riedel R, et al. Polymer-derived ceramics: 40 years of research and innovation in advanced ceramics[J]. Journal of the American Ceramic Society, 2010, 93(7):1805~1837
11 Xue Fengdan, Zhou Kechao, Wu Ning, et al. Porous SiC ceramics with dendritic pore structures by freeze casting from chemical cross-linked polycarbosilane [J]. Ceramics International, 2018, 44(6): 6293~6299
12 He Zhao, Lian Pengfei, Song Yan, et al. Protecting nuclear graphite from liquid fluoride salt and oxidation by SiC coating derived from polycarbosilane [J]. Journal of the European Ceramic Society, 2018, 38(2):453~462
13 Kaur S, Riedel R, Ionescu E. Pressureless fabrication of dense monolithic SiC ceramics from a polycarbosilane [J]. Journal of the European Ceramic Society, 2014, 34: 3571~3578
14 Li Houbu, Zhang Litong, Cheng Laifei, et al. Effect of curing and pyrolysis processing on the ceramic yield of a highly branched polycarbosilane [J]. Journal of Materials Science. 2008, 44:721~725
15 Huang Muhe, Fang Yunhui, Li Ran, et al. Synthesis and properties of liquid polycarbosilanes with hyperbranched structures [J]. Journal of Appllied Polymer Science, 2009, 113: 1611~1618
16 Luo Zheng, Zhou Xingui, Yu Jinshan, et al. High-performance 3D SiC/PyC/SiC composites fabricated by an optimized PIP process with a new precursor and a thermal molding method [J]. Ceramics International, 2014, 40: 6525~6532
17 Kissinger E D. Reaction kinetics in differential thermal analysis [J]. Analytical Chemistry, 1957, 29(11): 1702~1706
18 Ozawa T. Initial kinetic parameters from thermogravimetric rate and conversion data [J]. Bulletin of the Chemical Society of Japan, 1965, 38(11): 1881~1886
19 Crane L W, Dynes P J, Kaelble. Analysis of curing kinetics in polymer composites [J]. Journal of Polymer Science: Polymer Letter Edition, 1973, 11(8): 533~540
20 Frost R L, Cash G A, Kloprogge J T. ‘Rocky Mountain leather’, sepiolite and attapulgite an infrared emission spectroscopic study [J]. Vibrational Spectroscopy, 1998, 16(2): 173~184
21 陈曼华,陈朝辉,肖安. 聚碳硅烷/二乙烯基苯粘度特性研究[J]. 热固性树脂,2004,19(1):8~9
Curing and Ceramization of A Liquid Polycarbonsilane Precursor
Zhu Shibu Zhang Qiang Meng Xiangli Yang Xing Yan Liansheng Zhang Xiaohu Cui Hong
(Xi’an Aerospace Composites Research Institute, Xi’an 710025)
The ceramization and curing behaviors of liquid polycarbosilane (LPCS) precursor for SiC ceramic were investigatedby differential scanning calormetry (DSC) and thermogravimetric analysis (TGA). The structures and morphology of ceramic products were characterized by fourier transform infrared spectrometry (FTIR), X-ray diffraction (XRD) and scanning electron microscope (SEM). The kinetic parameters of this precursor were calculated by Kissinger, Ozawa and Crane equations, and the results were summarized as following: apparent activation energy (a) was 96.84kJ/mol and reaction order was 0.94. The mass loss for LPCS mainly occurred in 400~800℃, in which organic groups decreased and polymer-to-ceramic transformation completed. The XRD results indicated that the ceramic product maintained amorphous structure below 1000℃, and ceramics started to crystallize above 1500℃, resulting in the formation of β-SiC structure. LPCS possessed relative low temperature viscosity (0.25Pa·s) and well stability in air, so LPCS precursor was suitable for preparing SiC composite by means of PIP process.
LPCS precursor;curing kinetics;ceramization;SiC ceramic

国防基础科研项目(JCKY2017203C041)。
朱世步(1986),博士,材料学专业;研究方向:陶瓷基复合材料制备与性能。
2019-05-22
猜你喜欢
纺织学报(2023年9期)2023-10-31 08:27:08
少先队活动(2022年10期)2022-12-09 08:59:34
小哥白尼(趣味科学)(2022年7期)2022-09-20 06:07:34
陶瓷学报(2020年5期)2020-11-09 09:22:48
纺织科学与工程学报(2020年1期)2020-06-12 09:14:42
肿瘤预防与治疗(2020年1期)2020-03-07 10:47:56
酒·饮料技术装备(2018年1期)2018-04-28 09:08:56
资源再生(2017年3期)2017-06-01 12:20:59
核科学与工程(2015年2期)2015-09-26 11:57:24
上海塑料(2015年3期)2015-02-28 14:52:05
