
2-氨基苯腈合成拉帕替尼药物中间体工艺
2019-07-02 12:12:36王艳戎李子成
实验室研究与探索 2019年6期
张 宏, 王艳戎, 罗 兰, 李子成
(1.四川化工职业技术学院 化学工程系,四川 泸州 646099;2.泸州职业技术学院 科技产业处,四川 泸州 646000;3.四川大学 化工学院, 成都 610065)
0 引 言
N-(3-氯-4-(3-氟苄氧基)苯基)-6-溴喹唑啉-4-胺是靶向抗肿瘤药物拉帕替尼(Lapatinib)的重要中间体之一[1-2]。Lapatinib是由GSK公司开发研制,经全球众多研究机构近年来的努力,拉帕替尼的合成工艺正向着绿色、环保、高效、可循环的方向发展[3]。本文通过优化文献方法,以2-氨基苯腈为起始原料,经碘代、缩合、Dimroth重排闭环反应,制备N-(3-氯-4-(3-氟苄氧基)苯基)-6-溴喹唑啉-4-胺,其结构经LC-MS、1H-NMR确定,同时对相关碘代试剂的反应条件进行评价,增强2-氨基苯腈碘化效果,提高2-氨基-5-碘苯腈收率。
1 实验部分
1.1 主要设备和试剂
设备:LCMS-液质联用仪(Shimadzu 2010A,岛津)。
试剂:2-氨基苯腈(99.5%,上海氟德化工);间氟苄基氯(99.5%,阿法埃莎(中国)化学有限公司);2-氯-4-硝基苯酚(99.0%,百灵威科技有限公司);一氯化碘(99.0%,上海迈瑞尔化学技术有限公司);N,N-二甲基甲酰胺二甲缩醛(99.3%,北京欣赛维化学科技有限公司);薄层层析用硅胶(GF254,青岛海洋化工厂);其余试剂均为阿拉丁试剂,使用前经过二次除水(必要时)。
1.2 N-(3-氯-4-(3-氟苄氧基)苯基)-6-溴喹唑啉-4-胺合成路线
Williamson醚化反应是该系列反应的关键步骤之一。相关研究显示,化合物(2)3-氯-4-(3-氟苄氧基)苯胺是以间氟苄醇或卤代间氟苄基苯为初始原料,首先在碱性体系下,以KI为催化剂,通过Williamson缩合成醚。碱性试剂可以选用有机碱(三乙胺、二异丙基乙胺、二乙胺)和无机碱(Na2CO3、NaHCO3、KHCO3、K2CO3),以收率作为评价指标的单因素实验中发现,K2CO3-KI体系下的产物(1)收率最高,副反应较少,因此碱性试剂选用K2CO3。溶剂体系可以选用DMF、CH3CN、丙酮、乙酸乙酯。但DMF沸点较高,常规蒸馏和减压旋蒸都不易回收循环利用;CH3CN体系虽成醚效果较好,但毒性较大、成本昂贵;乙酸乙酯体系的成醚效果较差;丙酮体系对无机碱性催化体系(K2CO3-KI)的溶解效果优于乙酸乙酯,成醚效果较好,且价格便宜,沸点低,回收精制后可循环使用[4-5]。
随后将硝基(—NO2)加氢还原成胺基(—NH2)。还原试剂可采用H-试剂(NaH)、催化加氢(Pd/C)、金属还原(Fe粉/NH4Cl)等多种方法,由此可见还原剂的选择及活性是化合物(2)合成的关键点。NaH易燃且不易保存;贵金属Pd/C催化剂成本较高,并且需要肼盐做供氢体;Fe/NH4Cl还原反应后会产生大量铁泥,后处理工艺繁琐。为此,选择活性较高、易后处理的Zn/NH4Cl还原硝基的体系。
从经济高效、适宜工业生产的角度出发,结合众多学者研究成果,化合物(2)的合成途径[6-7]:以2-氯-4-硝基苯酚和间氟苄基氯为初始原料,K2CO3-丙酮体系下进行Williamson成醚反应生成化合物(1)3-氯-4-(3-氟苯甲氧基)硝基苯,在Zn/NH4Cl还原体系下,由化合物(1)制备得到化合物(2)。
化合物(4)合成途径:以2-氨基苯腈为原料,ICl为碘化剂合成化合物(3)2-氨基-5-碘苯腈,于DMF-DME体系下生成化合物(4)N′-(4-碘-2-氰基苯基)-N,N-二甲基甲酰胺[9-10]。
一般而言,碘代过程通常使用高价碘代试剂(ICl、N-碘代丁二酰亚胺、碘乙酸等),无需添加额外的助氧化剂,但碘化过程收率并不理想,碘代试剂通常具有高毒、高腐蚀性,对环境弊端较大。根据绿色环保的化学理念,众多研究者对碘代-催化体系、助氧化剂、溶剂进行多方面的探讨[11,13],如I2-H5PV2MO10O40-O2/CH3CN-HOAc碘化体系;KI(KIO3)/NaNO2/H2SO4-CH3CN碘化体系;I2-H2O2-磺化MCM-41介孔分子筛/HCl碘化体系;NH4I/H2O2/PhMe碘化体系等。在以ICl为碘代试剂进行反应的同时,对I2-O2/Zn(NO3)2·6H2O/H2O碘化体系,I2/NO2/乙酸乙酯-乙腈碘化体系,NH2I/H2O2碘化体系进行评价(以2-氨基-5-碘苯腈的转化率、选择性为主要指标)。
化合物(2)、(4)通过Dimroth 重排闭环反应,得到目标产物化合物(5),合成过程无SOCl2、POCl3、水合肼等严重污染的毒性物质,更加安全环保,适宜一锅法反应。所设计的合成途径如图1所示。
1.3 HPLC检测方法
色谱柱Ultimate®XB-C18;流动相[12]:V(pH4.0乙酸铵缓冲液∶CH3CN)=70∶30;流速1.0 mL/min;检测波长260 nm;柱温35 ℃;进样量15 μL。
2 合成方法与结果
2.1 化合物(1)的制备
在150 mL三口烧瓶中,加入2.60 g(15 mmol)2-氯-4-硝基苯酚,2.85 g(18 mmol)K2CO3,0.30 g(1.8 mmol)KI,75 mL丙酮,开启搅拌,15 min后加入2.16 g(15 mmol)间氟苄基氯,加热回流,TLC点板监控反应进程(展开体系V(乙酸乙酯∶石油醚)=1∶2),6.5 h后停止加热,自然降温过滤,丙酮洗涤滤饼3次(20 mL/次),合并滤液,减压蒸干溶剂,真空干燥,得到浅黄色固体4.01 g,HPLC检测纯度95.2%,收率92.0%。LC-MS:283[M+H]+,1H-NMR (DMSO,300 MHz):8.36(d,J=2.6 Hz, 1H), 8.25(J=9.2 Hz, 2.7 Hz, 1H), 7.42(m, 2H), 7.33(m, 2H), 7.24(t, 1H), 5.43(s, 2H)。
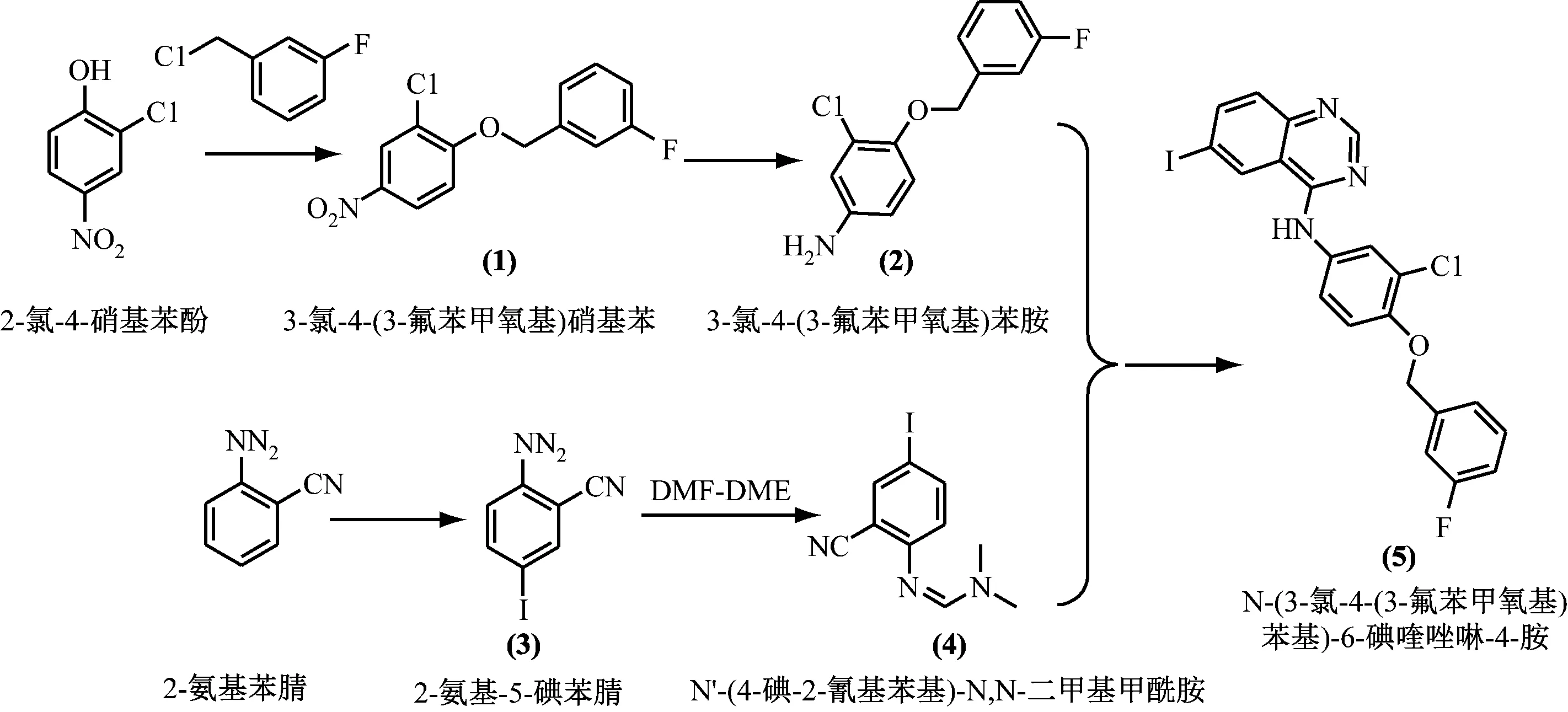
图1 N-(3-氯-4-(3-氟苯甲氧基)苯基)-6-碘喹唑啉-4-胺合成路线
2.2 化合物(2)的制备
Zn粉活化:称取10 g未活化锌粉置于50 mL 18 mol/L的盐酸溶液中,玻璃棒快速搅拌3 min后,混合液转移至G3砂芯漏斗内抽滤,蒸馏水洗涤滤饼至中性,CH3CH2OH洗涤2次,将滤饼置于真空干燥箱,25 ℃干燥4 h。活化后的Zn粉不宜长时间保存,一般使用前取相应量进行活化。
化合物(2)制备:1.2 g NH4Cl溶于少量水中,与50 mL乙醇混合后在搅拌条件下加入7.2 g活化后的Zn粉,用10%的盐酸调节体系pH至4,加热至45 ℃,搅拌30 min后,分3次(1.00 g/次)加入化合物(1),加热回流,TLC点板监测反应进程,2~3 h即可完全反应。趁热过滤,乙醇洗涤滤饼3次(20 mL/次),合并滤液,50 mL饱和食盐水反洗有机相,CH2Cl2-乙酸乙酯萃取,收集有机相,减压旋干,固相真空干燥,得到黄色固体2.48 g,HPLC检测纯度93.6%,收率86.9%。LC-MS:253[M+H]+;1H-NMR (DMSO, 300 MHz): 7.47~7.35(m, 1H), 7.29~7.24(m, 2H), 7.18~7.12(t, 1H), 6.93(d,J=8.6 Hz, 1H), 6.64(d,J=2.7 Hz, 1H), 6.49~6.44(J=8.7Hz, 2.5Hz, 1H), 5.08(s, 2H), 4.95(s, 2H)。
2.3 化合物(3)的制备
2.3.1 ICl-HOAC碘代体系
150 mL烧瓶内加入3.54 g(30 mmol)2-氨基苯腈完全溶于50 mL HOAc。将5.00g(30.8 mmol)ICl溶于20 mL HOAc中,在N2保护下,通过恒压滴液漏斗缓慢滴入烧瓶内,室温搅拌,4.5 h停止反应,向反应液中加入150 mL饱和食盐水,过滤,蒸馏水洗涤滤饼3次。滤饼于真空干燥箱干燥后,苯/石油醚(V=1∶3)体系下进行重结晶,得到3.92 g片状晶体,纯度90.3%,转化率48.2%。1H-NMR (DMSO, 300 Hz):7.71(d,J=2.2 Hz, 1H), 7.55(dd, 1H), 6.65(d,J=8.7 Hz, 1H), 6.24(s, 2H)。
2.3.2 I2-O2/Zn(NO3)2·6H2O/H2O碘化体系(Ⅰ)
150 mL烧瓶内加入30 mmol 2-氨基苯腈,5 mmol Zn(NO3)2·6H2O,15 mmol单质I2,60 mL H2O。首先在N2保护下[14-15],搅拌加热至有回流产生,将N2切换至O2-N2(体积比4∶1)混合气体,反应过程中通过HPLC对化合物(3)的转化情况进行跟踪分析。反应结束后,加入适量6%硫代硫酸钠水溶液进行反应淬灭,反应液中加入150 mL饱和食盐水,过滤,将滤饼溶于HOAc中,适量蒸馏水反洗有机相,分液,将HOAc旋蒸脱除,苯/石油醚(V=1∶3)体系下进行重结晶片状结晶,化合物(3)的转化率、目标产物选择性随反应时间的变化见表1。由表1数据可知,0~10 h内化合物(3)的转化率快速增长至44.7%,10~20 h化合物(3)转化率增长幅度降低,随着反应时间的进行,副反应增多,产物的选择性逐步降低。此反应体系与ICl-HOAc碘化体系对比,化合物(3)转化率没有明显提升,反应时长增加约4.3倍,故此碘化体系不太适用于2-氨基苯腈的碘化反应。
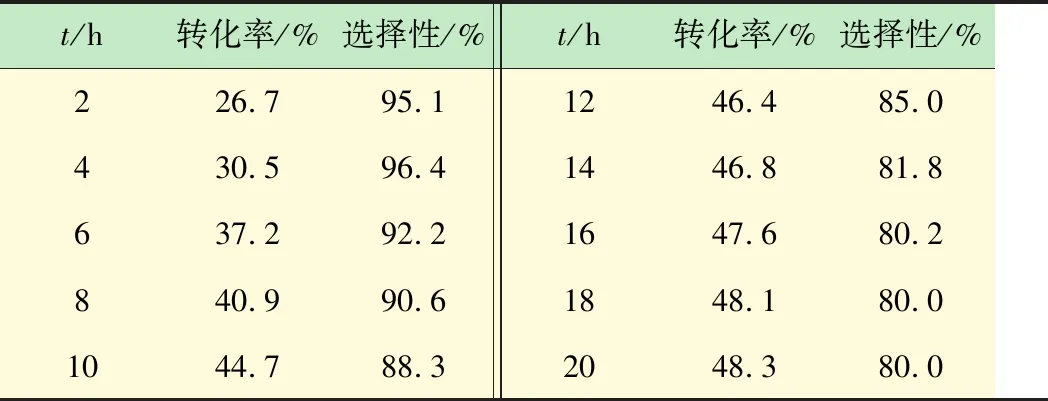
表1 碘化体系(Ⅰ)反应情况
O2作为助氧化剂与I2进行碘化反应,其机理可能是O2/Zn2+/硝酸根协同促使单质I2形成I8+高价碘活性中间体[16],如图2所示。
2.3.3 I2/NO2/乙酸乙酯-乙腈碘化体系(Ⅱ)
为便于实验操作和NO2计量,从安全角度出发,将NO2通入(质量分数)15%NaOH溶液中,制备6.5 mol%的氮氧化物钠盐,进行后续的碘化反应。
2NO2+2NaOH⟹NaNO3+NaNO2+H2O

图2 碘化过程机理推测
向250 mL带聚四氟乙烯内衬的高压反应釜内加入30 mmol 2-氨基苯腈,15 mmol单质I2,80 mL 6.5 mol%的NO2-NaOH溶液,30 mL CH3CN-HOAc混合溶剂(体积比3∶1)。反应釜首先用N2置换3次,之后通入N2至压力达到0.5 MPa,搅拌加热至有回流产生,将N2切换至O2-N2(体积比4∶1)混合气体,反应过程中通过HPLC对化合物(3)的转化情况进行跟踪分析,化合物(3)的转化率、选择性随反应时间的变化见表2。

表2 碘化体系(Ⅱ)反应情况
由表2数据可知,反应8 h化合物(3)的转化率达到49.9%,反应16 h后转化率达到58.2%,虽然耗时较高,但相比于ICl体系,转化率增长了10%左右。同样,反应时间的延长,副反应增多,生成产物的选择性逐步降低。从绿色环保反应角度出发,碘化体系(Ⅱ)的环境负荷低于ICl-HOAc。
2.3.4 NH2I/H2O2碘化体系(Ⅲ)
150 mL烧瓶内加入30 mmol 2-氨基苯腈,80 mL甲苯。开启搅拌至氨基苯腈完全溶解,在加入30 mmol NH2I,在N2保护下,加热至产生回流,将300 mmol H2O2通过恒压滴液漏斗匀速滴入烧瓶内,滴加完毕后,继续在N2保护情况下进行碘化反应。反应过程中通过HPLC对化合物(3)的转化情况进行跟踪分析,其转化率、选择性随反应时间的变化见表3。
由表3数据可知,反应4h化合物(3)的转化率仅有9.4%,反应66h后转化率达到47.8%,与ICl-HOAc体系相似,反应78h,化合物(3)的转化率基本稳定在63%左右,此体系碘化反应时间很长,相比于ICl体系,转化率增长了15%左右,但过程的副反应较少,90 h的产物选择性还在90%以上。就绿色化学而言,碘化体系(Ⅲ)的环境负荷显著低于ICl-HOAc,碘化反应中催化剂种类较少,反应条件温和易达到,如果能有效降低碘化反应所需时间,该路线基本符合工业化大生产的要求。
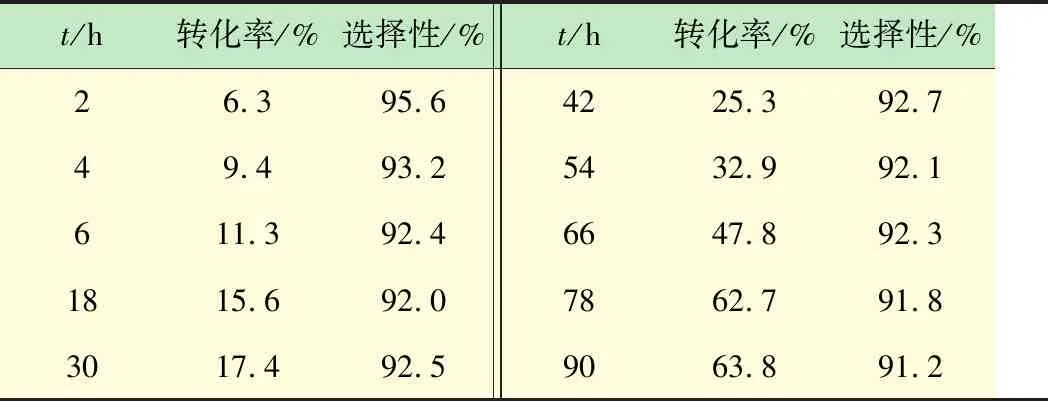
表3 碘化体系(Ⅲ)反应情况
2.4 化合物(4)的制备
将2.20 g化合物(3)加入至6 mL N,N-二甲基甲酰胺二甲基缩醛(DMF-DMA)中,加热回流1.5 h,减压蒸出过量的DMF-DMA,得到化合物(4)直接进行下一步Dimroth重排闭环反应。
2.5 化合物(5)的制备
将化合物(4),2.0 g(7.97 mmol)化合物(2),20 mL HOAc加热回流1.5 h后,停止加热,自然降温。60 mL饱和食盐水反洗反应液,CH2Cl2-乙酸乙酯萃取,合并有机相,旋蒸除溶剂,固相经真空干燥,得到黄色固体3.58 g,HPLC检测纯度91.0%,收率81.4%。LC-MS:507[M+H]+;熔点:222.0~224.0 ℃,1H-NMR(DMSO, 300 Hz):9.86(s, 1H), 8.97(s, 1H), 8.65(s, 1H), 8.16(d,J=8.7 Hz, 1H),8.08(s,1H),7.79(d,J=9.0 Hz,1H),7.57(d,J=8.6 Hz, 1H), 7.48(m,1H), 7.35~7.27(m, 3H), 7.21(t, 1H), 5.30(s, 2H)。
3 结 语
本文以2-氯-4硝基苯酚和间氟苄氯通过Williamson缩合成醚,Zn/NH4Cl还原制备化合物(2),反应的总收率为79.9%。此步骤的还原体系Zn/NH4Cl,Zn粉经过活化处理,提高了还原效率。该体系较Pd/C体系、Raney Ni-水合肼体系、NaH体系相比,在尽量保证产品收率的前提下,降低生产成本,未经活化的Zn/NH4Cl易于长期保存。
以ICl为碘化剂,对2-氨基苯氰进行进行碘代反应合成化合物(3),化合物(3)在DMF-DME体系下进行胺-醛缩合得到化合物(4),化合物(4)与化合物(2)在HOAc体系下进行Dimroth 重排闭环反应制得目标产物化合物(5),单步收率84.1%。该过程为拉帕替尼制备过程中的Suzuki偶联反应提供原料,基本符合投入工业化生产的要求。
然而,反应过程中应用的碘代剂ICl对环境有一定的负荷,遇水易分解,碘代效果不理想,故由2-氨基苯氰制备化合物(5)反应总收率仅为40.5%。因此对三种新碘化体系进行评价:I2-O2/Zn(NO3)2·6H2O/H2O碘化体(Ⅰ)系,I2/NO2/乙酸乙酯-乙腈碘化体系(Ⅱ),NH2I/H2O2碘化体系(3)。碘化体系(Ⅰ)不适合2-氨基苯氰的碘化反应;碘化体系(Ⅱ)的产物转化率有所提升,但选择性不理想;碘化体系(3)反应耗时较长,但产物的转化率和选择性较为理想,反应90 h,2-氨基苯腈转化率63%,选择性91.2%,较为符合绿色化学的要求。
猜你喜欢
新疆有色金属(2022年2期)2022-11-22 12:38:35
机械工业标准化与质量(2022年6期)2022-08-12 02:07:48
现代畜牧科技(2021年9期)2021-10-13 06:38:40
中国调味品(2017年2期)2017-03-20 16:18:13
中学生数理化·高二版(2016年5期)2016-05-14 13:19:33
精细石油化工(2015年3期)2015-12-14 09:07:42
中学化学(2015年2期)2015-06-05 07:18:13
无机盐工业(2015年2期)2015-02-17 07:05:58
中国药理学通报(2014年2期)2014-05-09 08:22:22
化工生产与技术(2014年3期)2014-02-27 13:41:42
