
天然产物中五元杂环生物合成过程的研究进展
2019-06-16 10:45张怡轩侯少阳章朦玥吴莹莹
微生物学杂志 2019年6期
张怡轩, 侯少阳, 章朦玥, 吴莹莹
(沈阳药科大学 生命科学与生物制药学院,辽宁 沈阳 110016)
五元杂环是一类含有除碳以外杂原子的五元环有机化合物。五元杂环分为含有一个杂原子的杂环和含有两个杂原子的杂环,含有一个杂原子的五元杂环有呋喃、噻吩、吡咯,含有两个杂原子的五元杂环有吡唑、咪唑、恶唑、异恶唑和噻唑[1]。五元杂环广泛存在于肽类[2-4]、生物碱类[5]及核苷类似物[6]等多种活性天然产物中。由于五元杂环化合物与核苷酸结构类似,具有形成氢键的特性,含有五元杂环的天然产物通常具有较好的生物活性,在抗菌[7]、抗炎[8]、抗病毒[9-10]、抗疟[11]、预防癌症[12]及免疫抑制[13]等方面均有深入研究。根据现有文献报道,细菌和真菌等微生物中五元杂环的前体可以是天然氨基酸[14-15]、乙酸[13]和乙酰辅酶A[16]等多种形式。早在1960年,Moyed等[17]提出了产气杆菌(Bacillusaerogenes)中组氨酸咪唑环的生物合成机制,是首个提出的五元杂环生物合成的成环机制;1970年,O′Donovan和Forde提出了南非醉茄(WithaniasomniferaDunal) 中以二氨基丁酸为底物的吡唑环成环机制[15];1996年,Kawamura等[18]提出了小四孢菌(MicrotetrasporaspiralisMI178-34F18)中以L-脯氨酸为底物的吡咯环成环机制,是首个提出的以氨基酸为底物生物合成五元杂环的成环机制;随后,Thomas等[19]于2002年详细阐述了霍乱弧菌(Vibriocholerae)中以L-脯氨酸为底物,经NRPS合成途径合成吡咯环;2001年,噻吩的生物合成过程经Margl等在孔雀草(Frenchmarigold)中推测得到[16];恶唑和噻唑在生物合成中的成环过程也相继被发现[12,20-22]。随着酶学和结构生物学的飞速发展,使得与五元环成环相关酶的特性和结构得到逐步解析[23-30],我们也能更加直观地认识五元环的成环过程。但是,部分五元环成环机制仍没有被完全阐明,如NRPS生物合成途径中恶唑和吡喃环的合成。五元杂环环化酶的底物通常分子量较小,在进行酶学特性实验和结构生物学分析时较为困难,因此对应酶学研究仍停留在假设阶段[31-33]。本文将从吡咯、恶唑和噻唑三种五元杂环入手,分别综述其在生物体内合成途径中的环化机制。
1 吡咯环的生物合成
吡咯环的生物合成底物可以为天然氨基酸、丙酮酸或乙酸等,成环方式多种多样,可直接由天然环脱氢氧化,也可以由小分子底物经过酶催化环化甚至可由底物经分子重排成环。
1.1 灵菌素中吡咯环生物合成过程
灵菌素(prodigiosins)是一种红色的天然产物,可由多种革兰阳性菌或革兰阴性菌产生[4,34-37],因其分子结构中有3个吡咯环,所以又名三聚吡咯。灵菌素的生物合成过程在γ-变形杆菌Serratiamarcescens中首次被发现[38],随后在假交替单胞菌Pseudoalteromonasrubra中解析[39]。灵菌素由脯氨酸、甘氨酸、丝氨酸和几个二碳单位合成,灵菌素生物合成基因簇由两个酶催化途径组成的单向多顺反子组成[40],两个酶分别催化合成2-甲基-3-n-戊烷基-吡咯(MAP)和4-甲氧基-2,2′-吡咯-5-甲醛(MBC)。如图1所示,MAP生物合成途径由pigB、pigD和pigE构成。首先,PigD在辅酶硫胺素焦磷酸(TPP)存在下催化丙酮酸形成辛烯酸,PigE催化其环化形成2-甲基-3-n-戊烷基-二氢吡咯(H2MAP),PigB催化H2MAP氧化形成MAP;MBC生物合成途径共包含7个基因(pigA、pigF~J、pigL和pigM),4′-磷酸泛酰巯基乙胺基转移酶(PigL)将PigG的酰基载脂蛋白(PCP)结构域由apo状态活化到holo状态;PigI在ATP的作用下催化L-脯氨酸的L-脯氨酰基团转移形成L-脯氨酰-S-PCP中间体,PigA将中间体氧化,形成吡咯基-2-羧基-S-PCP,由此第一个吡咯环形成;PigH的ACP结构域发生磷酸化为丙二酰辅酶A提供结合位点,结合的丙二酰辅酶A随后进行脱羧与连接在PigJ上的吡咯-2-羧基硫代酯发生缩合,在PigH上形成吡咯-β-酮硫代酯,PigH能催化丝氨酸和吡咯-β-酮硫代酯缩合形成4-羟基-2,2′-吡咯-5-甲醇[40-41],经过PigM、PigF和PigN催化氧化及甲基转移,最终形成MBC;PigC利用MAP和MBC在ATP的参与下将两个分子缩合形成灵菌素。
由上述可知,灵菌素中一个吡咯环由乙酸和乙酸铵等二碳单位经NRPS合成酶催化合成;另一个吡咯环由丝氨酸和脯氨酸经PKS合成途径共同环化而成;第三个吡咯环由脯氨酸经黄素依赖的脱氢酶氧化而来[36]。总的来说,灵菌素中吡咯环成环机制极具代表性,由NRPS-PKS杂合系统表达而来,涉及的酶反应较多,为五元环成环机制研究提供新视野。

图1 灵菌素生物的生物合成过程[36]
1.2 吡咯尼林中吡咯环的生物合成
吡咯尼林(Pyrrolnitrin,PRN),3-氯-4-(2-硝基-3-氯苯基)吡咯,是一种含有两个氯原子和一个硝基的苯基吡咯衍生物[42]。早在1964年,Arima等[43]就从假单胞菌(Pseudomonasspp.)中分离得到PRN类化合物;1966年,Lively等[44]研究表明,在假单胞菌(PseudomonasaureofaciensATCC 15926)中,PRN生物合成前体为L-色氨酸;随后Hammil等[45]的研究则表明含有D-色氨酸的培养基可使假单胞菌(Pseudomonasaureofaciens)的PRN高产。以色氨酸为底物经分子重排即得到吡咯尼林的吡咯环,具体生物合成途径如图2所示。首先色氨酸在黄素依赖的色氨酸-7-卤代酶(PrnA)的催化下生成7-氯-色氨酸,7-氯-色氨酸在单氯代氨基吡咯尼林合成酶(PrnB)催化下得到单氯代氨基吡咯尼林,由此,吡咯尼林中吡咯环形成。随后,黄素依赖的PrnC催化单氯代氨基吡咯尼林发生卤代反应,即在吡咯环上发生氯代,形成氨基吡咯尼林,最终在氨基吡咯尼林氧化酶(PrnD)作用下氨基被氧化为硝基,吡咯尼林最终合成。在吡咯尼林合成过程中,由天然氨基酸作为直接底物,吡咯环成环时不涉及其他小分子加入,仅仅依靠分子内重排形成吡咯环。

图2 吡咯尼林的生物合成途径[46-47]
2 噻唑环的生物合成
噻唑环在天然产物中广泛存在[4,12,21,25,48-49],现在已知的噻唑环化合物一般经过NRPS-PKS途径合成,合成底物一般为天然氨基酸和小分子碳单位。随着近年来酶学研究的快速发展,对噻唑合成酶的异源表达、分离纯化和特性的表征有了更深入的研究[50-51]。
2.1 埃博霉素中噻唑环的生物合成
埃博霉素(Epothilone)是一种具有抗肿瘤活性的内酯类化合物,由堆囊菌属(Sorangium)菌株产生[48]。埃博霉素类天然产物有A、B、C和D四种类似物,因D具有良好的抗肿瘤活性而备受关注[52-53],图3(a)即为埃博霉素D的结构。堆囊菌Sorangiumcellulosum中埃博霉素生物合成基因簇如图3(b)所示,epoA、epoB和epoC先后参与噻唑环的形成,图3(a)中化合物结构中标记的颜色对应图3(b)中基因簇的颜色,可以看出epoB直接参与噻唑环的环化;图3(c)描述了由丙二酰辅酶A合成噻唑环的具体过程,EpoA是epothilone生物合成的起始模块,首先装载丙二酰辅酶A,经过KSy结构域脱羧形成乙酰-S-EpoA,EpoB的Cy结构域具有双功能催化活性,首先催化乙酰基转移形成C-N键,接着在Cy结构域催化下形成噻唑烷基硫代酯中间体,EpoB的Ox结构域是黄素依赖的氧化酶,将噻唑烷氧化形成异芳基即噻唑环,接着传递给EpoC进行epothilone的后续合成。
Epothilone中噻唑环的合成底物为丙二酰辅酶A和半胱氨酸,经过EpoB的催化环化及氧化得到完整噻唑环。该合成过程的中间产物经过Chen等[25]利用HPLC和质谱等分析手段逐一分析,详细展示了epothilone中噻唑环的合成机制及相关酶的催化过程,为epothilone中噻唑环的形成提供理论和实验依据。
2.2 硫胺素中噻唑环的合成过程
硫胺素(维生素B1,vitamin B1)对于大多数微生物是必需的,在碳水化合物和氨基酸代谢中起着重要作用[54]。硫胺素在生物合成过程中分为噻唑和嘧啶两个独立过程,噻唑和嘧啶经过缩合形成硫胺素[55-56]。无论是真核生物还是原核生物均能合成硫胺素,现有文献常在细菌中讨论硫胺素嘧啶基的生物合成,在酵母等真核生物中讨论硫胺素噻唑环的合成[57]。噻唑环形成的分子机制在多种真核生物中得到深入解析,不同生物体内含有多种噻唑合成酶,比如酿酒酵母(Saccharomycescerevisiae)中的THI4p[58-59]、拟南芥(Arabidopsisthaliana)中的THI1[60-61]、米曲霉(Aspergillusoryzae)中的ThiA[62]。如图4(a)所示即为酿酒酵母中THI4p催化噻唑环合成的过程,噻唑环合成底物为NAD+、甘氨酸和半胱氨酸,THI4p催化NAD+和甘氨酸在单次成环反应中转化为ADT,THI4p的Cys205为噻唑环提供硫原子。

图3 埃博霉素生物合成简介
细菌生长同样需要硫胺素,多数细菌也可独立合成硫胺素中的噻唑环[55]。细菌中与硫胺素噻唑环合成有关酶的晶体结构如图4(b)所示,即ThiG和ThiS复合体。通过该晶体结构能更好地阐述细菌体内硫胺素噻唑环的合成,ThiG-ThiS复合体结构是点对称的8聚体结构,ThiG和ThiS在空间上较为接近使其能进行如图4 (c)所示反应,ThiG通过装载Deoxy-D-xylulose-5-phosphate,ThiS提供噻唑环的硫原子,经过脱水环化反应最终形成不带芳香性的五元环。随后,TenI催化脱氢形成具有芳香性的噻唑环,至此细菌中噻唑环合成。
硫胺素中噻唑环生物合成的原料为天然氨基酸和小分子碳单位,但不涉及NRPS或PKS合成过程,通常由噻唑合成酶直接环化脱水合成[58-59]。硫胺素中噻唑环的合成机制探讨的较为清楚,如图4(b)所示,噻唑环合成相关酶的晶体结构已经被确定,可以更加直观地认识噻唑环的环化过程。



图4 噻唑环生物合成简介
3 恶唑环的环化过程
和噻唑环类似,恶唑环也广泛存在于天然产物中[63]。恶唑环通常被认为由氨基酸作为底物直接合成(图5)[20,64]。已有文献报道了某些天然产物中的恶唑环是氨基酸来源的,如microncin[22,65]。但是仍有很多天然产物恶唑环的合成机制不清、前体不明,如oxazolomycin[31]、ulapulalides[66]等,这些化合物的恶唑环明显位于聚酮衍生的碳链内部,因此其恶唑环可能不是由氨基酸作为前体直接合成。
3.1 Microncin中恶唑环的生物合成
Microncin是一类原核核糖体和翻译后修饰肽(prokaryotic ribosomally synthesized and posttranslationally modified peptides,RiPPs),其恶唑环直接来源于半胱氨酸和丝氨酸残基[22]。如图5所示,microncin中恶唑环或噻唑环由B、C、D三蛋白复合体识别前导肽催化合成五元杂环。合成过程总体分为两步,首先蛋白C和D利用一分子ATP催化直链肽段环化,形成不具有芳香性的五元杂环;然后FMN依赖的蛋白B催化不具有芳香性的五元杂环脱水最终形成具有芳香性的五元杂环。图5(c)所示为蛋白D催化环化形成五元杂环的具体过程,即蛋白D利用ATP磷酸化肽段残基中的羰基氧磷酸化,使其脱水并完成环化反应。图5(d)中列举了其他含有恶唑环的RiPPs,可以看出由直链多肽合成五元杂环,每个生物合成基因簇中的生物合成酶较为类似,均含有一个前体肽解码酶和恶唑环合成酶(环化脱水酶和脱氢酶)直接由直链氨基酸残基经过环化脱水和氧化脱氢得到芳香恶唑环。
3.2 Oxazolomycins中恶唑环的合成过程
Oxazolomycins(OZMs)类化合物可由多种放线菌产生,近年来化合物的种类不断增多,并且表现出重要的抗菌、抗肿瘤和抗艾滋病病毒(HIV)活性[33,67-69]。Oxazolomycin A属于聚酮类化合物,由PKS-NRPS杂合基因簇合成,包含一个恶唑环,现已从多株链霉菌中分离得到[31-33,69]。如图6列举的OZMs类化合物生物合成的部分基因簇,虽然这类化合物的生物合成基因簇是已知的,但恶唑环的合成机制始终没有得到解析。2015年,Zhou等提出了两种由CongE催化的环化脱水形成恶唑环过程,图6(e)机制(1)中CongE通过ATP中焦磷酸盐的转移,来激活酰胺基氧磷酸化促进恶唑环的形成,该机制已被报道有助于恶唑环的形成[32];机制(2)中,ATP通过激活另一个酮基完成恶唑环的合成。与microncin中蛋白B、C和D的催化机制不同,CongE中没有氧化还原中心,无法进行与microncin中蛋白B相似的氧化反应,因此图6(e)的反应机制存在一定缺陷。本课题组通过对oxazolomycins类化合物基因簇的进化分析,进行恶唑合成相关酶的异源表达、纯化及体外反应工作,提出了新的OZMs类化合物中恶唑环化机制(待发表)。
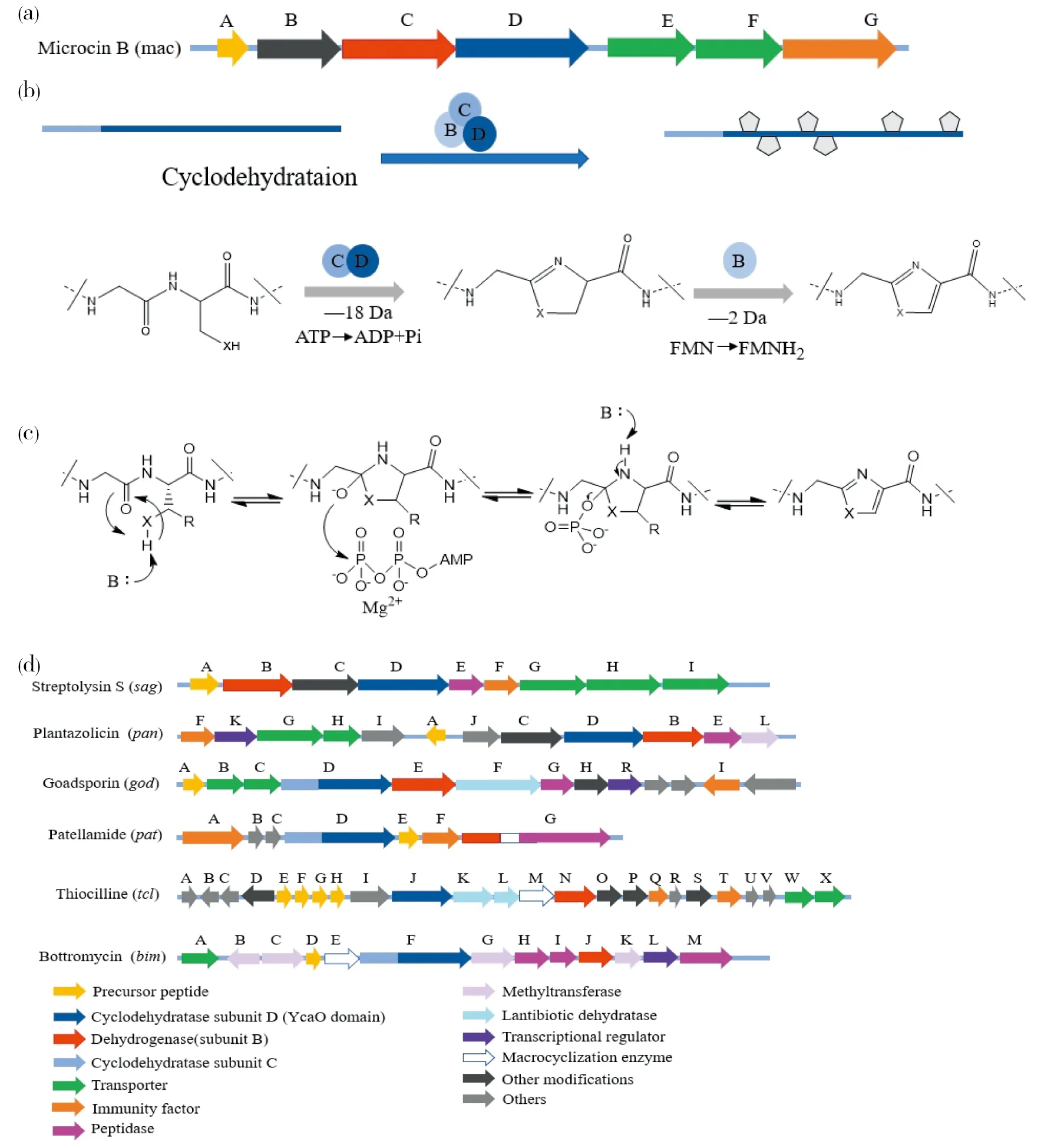
图5 RiPPs中噻唑环生物合成简介

图6 Oxazolomycins类化合物中恶唑环生物合成基因簇及成环机制简介
4 展 望
来自分子遗传学、结构生物学和生物化学领域的研究者使天然产物中五元杂环的生物合成过程和机制逐步清晰,五元杂环可由单酶催化,也可由多酶协作催化合成。这不仅可以解答重要的基础研究问题,更重要的是研究者可利用现有的五元杂环成环机制推测未知化合物的成环过程,为新化合物生物合成基因簇的表征和合成机制的推导提供思路。
就目前的研究成果看,多数五元杂环的合成过程研究仅停留在基因层面。由于五元杂环分子量较小,给结构生物学和酶催化机制研究带来了困难。随着组合生物合成的发展,如何选择合适的酶反应底物以更加合理地进行五元杂环环化酶反应中间体的捕获,给五元杂环的结构生物学研究提出了新的要求。此外,五元杂环在发挥生物功能时,通常嵌入到DNA中使DNA损伤,在含有五元杂环的天然化合物合成过程中,如何保卫自身细胞免受五元杂环化合物的损害还有待进一步研究。由此,五元杂环是某些天然产物具有良好生物活性的前提,含有五元杂环的化合物通常具有良好的抗菌、抗肿瘤和抗病毒等活性,可将五元杂环合成酶作为化合物合成模块,应用到组合生物合成中,为新化合物的发现及定向合成新化合物提供模块支持。
猜你喜欢
小作家报·教研博览(2022年11期)2022-04-02
材料科学与工艺(2022年1期)2022-03-11
中国瓜菜(2020年9期)2020-11-30
教育周报·教育论坛(2020年3期)2020-10-21
生物工程学报(2020年1期)2020-03-12
药学研究(2019年10期)2019-11-01
科技资讯(2018年16期)2018-10-26
科技信息·下旬刊(2018年8期)2018-10-21
农业与技术(2018年3期)2018-03-21
大众科学(2017年10期)2017-12-18
