
水稻硝态氮响应的酵母双杂交文库的构建与分析
2019-06-14 01:26洪广成刘石锋张汉马秦小健
生物学杂志 2019年3期
洪广成, 陈 倩, 刘石锋, 张汉马, 秦小健
(重庆师范大学 生命科学学院 植物环境适应分子生物学市重点实验室, 重庆 401331)
酵母双杂交技术是Fields等首先创立的,是目前研究蛋白质与蛋白质之间相互作用的主要方法[1-3]。其原理是利用酵母特异转录激活因子GAL4(galactose-regulated upstream promoter element, GAL )具有DNA 特异结合域( DNA-binding domain )与转录激活结构域( transcription-activating domain )两个彼此分离的结构域,两个结构域之间相互独立互不影响,但只有同时含有这两个结构域时才能转录激活特定的表达基因,具有高效、简便、高灵敏度等优点[4-7]。
水稻作为我国三分之二人口的主食,水稻产量的稳定与增收对于整个社会的稳定发展具有重要意义[8]。在过去的几十年中,人工合成氮肥的出现极大地提高了水稻的产量[9]。但由于过量施肥和不科学的施肥方式导致了水体富营养化、环境污染和温室效应等一系列生态问题的出现。因此在农业实际生产中, 大量施加的氮肥若不能被水稻高效吸收利用, 造成的资源损失和环境负面影响要比实际收益大得多[10-13]。
目前水稻产量受技术和养分的限制已到达瓶颈,需要从基因和其他方向做突破[14-15]。为了研究硝态氮响应条件下的相关水稻基因调控机理,本研究构建了水稻氮响应cDNA文库,为后续研究提供帮助。
1 材料与方法
1.1 材料
1.1.1 植物材料 籼型杂交稻恢复系“9311”系中籼水稻品种(OryzasativaL.spp.indicavar.9311)。
1.1.2 主要试剂 “Mate & Plate”文库构建试剂盒、PCR产物纯化试剂盒、SD/-Leu、SD/-Leu/-Trp 和 SD/-Ade/-His/-Leu/-Trp 购自Clontech 公司;质粒DNA提取试剂盒、胶回收试剂盒购自TIANGEN公司;TRIzol®Reagent 购自Life Technologies 公司。
1.2 方法
1.2.1 水稻幼苗的培养与处理
用改良后不含氮素的霍格兰营养液水培,以KNO3作为唯一氮源,在培养箱内先将已催芽的水稻种子放置于低氮(0.3 mmol/L)条件下培养15 d,再将其迅速转移至高氮(5 mmol/L)水平条件下培养4 h后迅速提取处理材料的总RNA以备后用。
1.2.2 水稻总RNA的提取与检测
将已处理的水稻组织放入加有液氮的研钵中研磨至粉状,用Trizol提取法提取水稻的总RNA,具体步骤可参考Trizol试剂说明书。试验所用的研钵经酒精灼烧,所用离心管、枪头等均经过RNase free处理过,样品摆放在冰面上,全程在超净工作台上操作,离心采用低温离心机,防止提取过程中总RNA发生降解,最后将分离纯化得到的总RNA用30 μL的DEPC水溶解。用BioDrop分光光度计测RNA浓度,并用浓度为 1% 的琼脂糖凝胶电泳检测提取的 RNA 质量。
1.2.3 RNA反转录cDNA与检测
建立反转录体系合成第一链 cDNA,以提取的水稻总RNA作为模板,CDS III(5′-ATTCTAGAGGCCGAGGCGGCCGACATG-d(T)30VN-3′)为引物。再以合成的第一链cDNA为模板,使用长距离 PCR (LD-PCR) 扩增 ds cDNA。用CHROMA SPIN+TE-400 纯化柱纯化扩增的 ds cDNA,除去200 bp以下的小片段,具体步骤参考说明书 PT4085-1(Clontech)。用浓度为1%的琼脂糖凝胶电泳检测纯化后的ds cDNA。
1.2.4 Y187酵母感受态制作
挑取划线平板上生长最快的Y187酵母单克隆菌株用YPDA培养液扩大培养,并用分光光度计检测菌液的OD600值,达到浓度后用1.1×TE/LiAc溶液制备建库所需的感受态酵母,具体操作步骤参考说明书 PT4085-1(Clontech)。
1.2.5 构建cDNA文库
用DMSO增强感受态酵母的细胞通透性,以高温变性后迅速低温处理的单链鲑鱼DNA为携带载体,用热激法将质粒载体pGBKT7-Rec和纯化处理的双链cDNA 共同转入感受态酵母细胞。用YPD Plus Medium恢复处理转化的酵母细胞后,再用15 mL 0.9%(W/V) NaCl溶液悬浮菌液,涂布于SD/-Leu培养基上,30℃倒置培养3 d后,用玻璃珠和冷冻液收集菌体,得到的酵母菌悬浮液即为构建的酵母双杂交cDNA文库;将菌液分装在1.5 mL无菌离心管中,-80℃低温保存,详细步骤参照说明书 PT4085-1(Clontech)。
1.2.6 cDNA文库转化率及文库大小的测定
从 15 mL 0.9% (W/V) NaCl 菌悬液中吸取100 μL菌液,稀释10倍、100倍后分别吸取100 μL稀释后的菌液涂布于 SD/-Leu 培养基上 30℃倒置培养 3 d,统计培养基上的单克隆菌落数计算转化效率。取10 μL收集在冷冻液中的菌液稀释102、104和106倍后,分别吸取100 μL涂布于SD/-Leu 培养基上30℃倒置培养3 d,统计平板上的单克隆菌落数计算文库滴度。
1.2.7 cDNA文库平均插入片段大小及重组率的计算
从计算转化效率的SD/-Leu 培养基上随机挑选20个单菌落扩大培养,提取酵母质粒DNA。以提取的质粒DNA为模板,pGADT7-Rec载体上游引物 Primer: 5′-CTATTCGATGATGAAGATACCCCACCAAACCCA-3′,下游引物 Primer:5′-GTGAACTTGCGGGGTTTTTCAGTATCTACGATT-3′进行PCR 扩增。扩增程序:95℃预变性3 min; 95℃变性 30 s, 58℃退火 30 s, 72℃延伸 3 min,30个循环; 72℃再延伸10 min; 最后12℃保存。反应结束后用浓度1%的琼脂糖凝胶电泳检测 PCR 产物。
1.2.8 cDNA文库平均插入片段的分析
从计算文库滴度的培养基中随机挑选10个单克隆,提取质粒后测序,并在NCBI上用Blast 功能对测序结果进行序列比对,寻找匹配序列。
2 结果与分析
2.1 总RNA 的提取
用Trizol提取法提取处理过的水稻材料9311的总RNA。将提取后的总RNA用浓度1%的琼脂糖凝胶电泳检测,结果显示28S及18S条带清晰,5S条带较弱(图1),RNA的完整性较好。经过分光光度计检测,浓度约 2 μg/μL ,表明 RNA的质量较好,可用于下一步文库的构建。
2.2 cDNA的合成与文库的构建
取 2 μL 提取的总RNA为模板,反转录合成第一链cDNA,再以合成的第一链cDNA为模板,用LD-PCR体系扩增出ds cDNA,经浓度1% 琼脂糖凝胶电泳检测显示,ds cDNA的电泳条带呈弥散状,大小集中分布在 500~3000 bp 范围内(图2)。将合成的ds cDNA与载体pGADT7-Rec混合共转化到感受态酵母菌Y187中得到cDNA文库。 收集cDNA文库的菌体,使用1.5 mL无菌离心管每管分装1 mL后存放于-80℃备用。

图1 总RNA电泳检测
Figure 1 The electrophoresis of total RNA detection
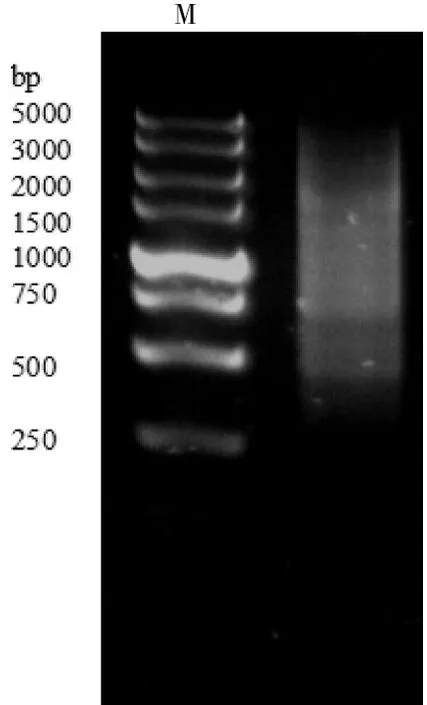
图2 ds cDNA 电泳检测
Figure 2 The electrophoresis of total ds cDNA detection
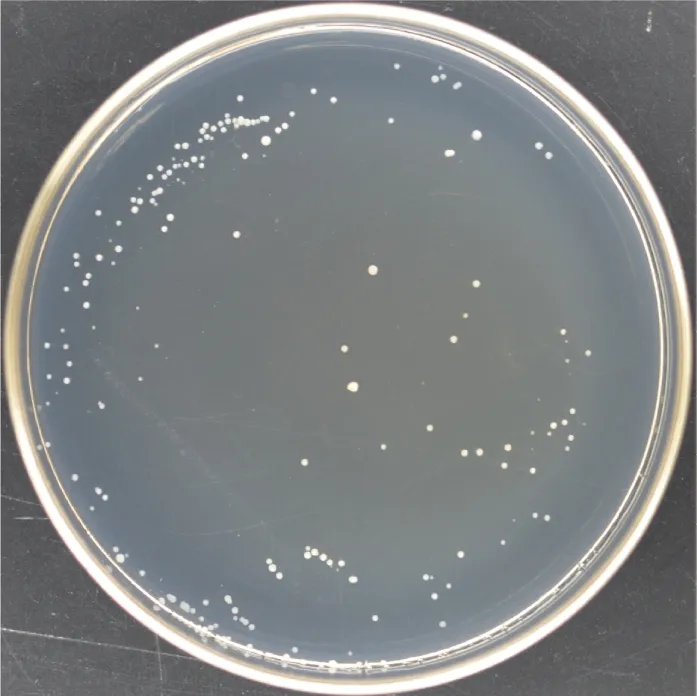
图3 计算文库转化效率SD/-Leu 培养基
2.3 cDNA 文库的转化效率及插入片段大小检测
文库转化完成后加入 15 mL 0.9% (W/V) NaCl溶液悬浮菌液,吸取10 μL悬浮后的菌液稀释100倍涂布于SD/-Leu 培养基上,30℃恒温箱倒置培养3 d(图3)。统计平板上的克隆数共215个,转化效率计算公式:平板上的单克隆数×稀释倍数×总转化混合物体积/涂布体积 /3 μg pGADT7-Rec,计算得到转换率为3.23×106/3 μg pGADT7-Rec满足实验需要的1×106/3 μg pGADT7-Rec。将收集在冷冻液中的 cDNA 文库混合均匀,吸取 10 μL菌液稀释100倍,再取10 μL稀释后的菌液稀释100倍,最后取10 μL稀释两次后的菌液稀释100倍得到3种不同浓度的文库,即原浓度的10-2、10-4和10-6。分别取100 μL稀释后的文库涂布于SD/-Leu 培养基上,30℃恒温箱倒置培养3 d(图4)。在稀释至10-6的平板上平均长出17个单克隆菌落,文库滴度(CFU/mL)= 单平板平均菌落数(17)/ 涂布体积(0.1 mL)×稀释倍数(106)=1.7×108cfu/mL,满足实验所需滴度 2×107cfu/mL。从转化效率的平板上随机挑取20个单菌落进行 PCR 检测只有1个未能扩增出相应的片段(图5),重组率达 95%; 插入片段长度介于500 ~1500 bp 之间,平均插入片段长度在1000 bp左右,文库质量较好,可用于后续互作蛋白的筛选。

A、B、C:稀释至10-2、10-4和10-6后的酵母菌悬液分别在文库 SD/-Leu 平板上的生长情况,用于计算文库滴度
图4 不同浓度的 cDNA 文库在 SD/-Leu 培养基上的生长情况
Figure 4 The growth of different concentrations cDNA library yeast on SD/-Leu medium
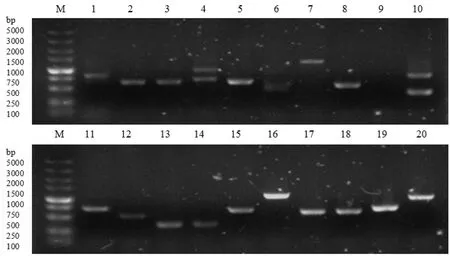
图5 cDNA 文库插入片段大小电泳检测
2.4 cDNA 文库插入片段分析
从文库中随机挑选10个克隆测序后,通过NCBI上Blast序列比对分析,共有9个克隆匹配到同源序列(表1)。其中除了2、6插入片段在序列比对时有部分碱基匹配错误,其他插入片段编码顺序均较匹配。
3 讨论与结论
cDNA文库的质量取决于文库的代表性与完整性[16-17],而完整性的体现需要总RNA的高质量提取,在提取过程中几乎未发生RNA的降解,并且RNA高度纯化无杂质。本试验所用的总RNA经过纯化处理后的电泳图片28S和18S条带清晰,表明总RNA的质量较高,可用于文库的构建。
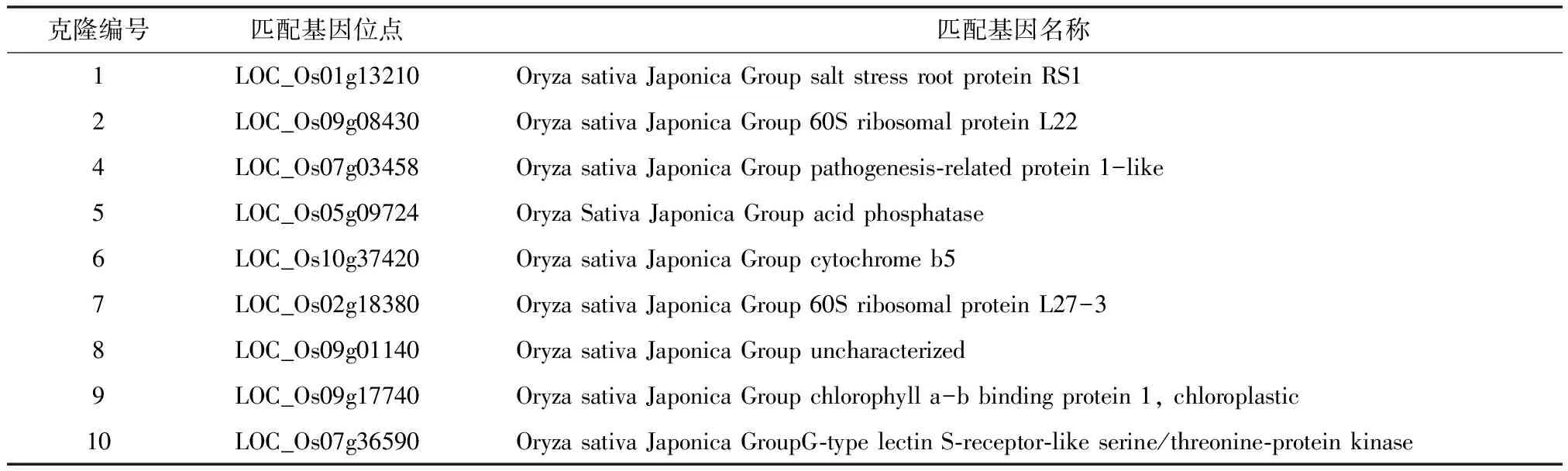
表1 测序匹配基因信息统计
本试验使用的是Clontech 公司“Mate & Plate”文库构建系统构建的水稻 cDNA文库。用 SMART 技术合成的 ds cDNA 不需要对总RNA中的mRNA进行分离纯化,因而避免了mRNA的降解损失;反转的ds cDNA再通过 CHROMA SPIN TE-400 柱子的纯化,去除小片段DNA,减少cDNA文库在整合外源片段时产生的不利因素。同时利用同源重组的方法将与 pGADT7-Rec 载体末端同源的ds cDNA 定向重组到pGADT7-Rec 载体上,不仅可以有效避免繁琐的酶切连接等步骤,同时也解决了连接的低效性和克隆的嵌合问题,从而保证了cDNA 文库整合外源 ds cDNA 时的方向性和完整性[18]。通过转化效率检测的结果达到3.23×106/3 μg pGADT7-Rec满足实验所需条件,保证了试验文库的完整性和代表性。文库的插入片段大小在1500 bp以下,说明大片段cDNA在同源重组时的效率较低,为以后的文库构建提供经验。
本试验构建的水稻9311氮响应条件下的cDNA酵母文库,cDNA文库滴度1.7×108cfu/mL,插入的片段大小在500~1500 bp之间,文库的质量符合筛选条件,可用于后续水稻氮吸收与利用关键基因互作蛋白筛选,为水稻氮素利用分子调控通路关键基因的挖掘提供帮助。
猜你喜欢
太原理工大学学报(2021年6期)2021-11-25
猪业科学(2021年3期)2021-05-21
中国现代医药杂志(2020年10期)2020-12-14
幽默大师(2020年10期)2020-11-10
海洋科学(2020年9期)2020-10-09
中华诗词(2019年1期)2019-11-14
中国海洋大学学报(自然科学版)(2019年7期)2019-05-21
中国海洋大学学报(自然科学版)(2019年7期)2019-01-04
猪业科学(2018年4期)2018-05-19
医学研究杂志(2015年3期)2015-06-10
