
基于微滴式和芯片式数字PCR技术的转基因克螟稻成分的定量检测
2019-05-21 11:59:52邓婷婷黄文胜葛毅强
食品科学 2019年8期
邓婷婷,黄文胜,葛毅强*,陈 颖,*
(1.中国农业大学食品科学与营养工程学院,北京 100083;2.中国检验检疫科学研究院,北京 100176;3.科技部中国农村技术开发中心,北京 100045)
随着转基因作物种植面积的日益扩大和着转基因食品飞速发展,许多国家都出台转基因标识标签制度,欧盟、韩国及日本的转基因标识阈值(质量分数)分别为0.9%、3.0%、5.0%[1-3],我国自2002年起实施《农业转基因生物标识管理办法》[4],明确规定在中国境内销售农业部许可并列入标识的转基因生物及其产品,必须进行转基因成分的标识。随着全世界标识制度的完善和定量标识制度的发展,建立转基因产品的检测技术尤其是精准定量检测技术将越来越重要。
目前国内外针对转基因成分定量分析最常用的方法是实时荧光聚合酶链式反应(polymerase chain reaction,PCR)法,该技术依靠不同浓度的标准样品建立标准曲线对未知模板进行定量分析,已比较成熟地用于转基因定值当中[5-8]。我国也发布了多项基于实时荧光PCR技术的转基因作物定量检测标准方法[9-10],其定量灵敏度在0.5%~0.1%之间。但是,实时荧光PCR技术用作定量时由于每次进行检测操作都需同时进行已知浓度标准品的扩增并制作标准曲线,校准品和样品间背景的不同及PCR扩增效率等因素都引入了偏差,导致其定量结果往往与实际值相差甚远[11-12]。此外,低拷贝数的目标DNA分子很难通过扩增检测到[13-15],因此在实际应用中局限性较多。
数字PCR是近年来发展起来的单分子DNA绝对定量检测技术,通过对样品DNA极度稀释,对每个微反应单独进行PCR扩增后采用终点法检测PCR产物,并依据泊松分布原理对靶基因定量,在不需要标准样品的情况下实现目标成分的绝对定量[16],有效避免了上述实时荧光PCR定量带来的扩增反应抑制物、PCR扩增效率差异及标准曲线等带来的定量偏差等问题,并可减少实时荧光PCR带来的基体效应[17]。数字PCR具有测量独立性与无需任何校准物的特点,也使得低丰度靶标分子的精准检测成为可能[13,18-19]。由于数字PCR定量更加准确灵敏,目前已经逐渐应用到转基因定量分析和低含量的转基因成分检测中。Fu Wei等[20]选取CaMV35s启动子和NOS终止子作为筛选转基因作物的通用元件,用数字PCR进行定量检测,并对MON810等9 种转基因品系进行特异性验证,结果获得检测限为0.1%这一低于欧盟规定阈值的结果,表明该方法特异性好、灵敏度高,适于转基因成分含量的精确定量。David等[21]利用微滴式数字PCR方法分别建立了两个多重定量方法,可对12 个品系的转基因玉米进行含量测定,其检出限、重复性和真实性均符合国际标准中GMO定量要求。其他研究也表明与实时荧光PCR方法相比,数字PCR定量方法定量更加准确灵敏[22-24]。
本研究基于微滴式和芯片式数字PCR平台,以抗虫转基因水稻品系克螟稻为研究对象,拟建立转基因水稻的精准定量分析方法,并与传统的实时荧光定量PCR结果进行比较,以期为食品和农产品中转基因成分的定量分析提供新的方法和借鉴。
1 材料与方法
1.1 材料与试剂
转基因水稻(Oryza sativa)LL62品系(大米样品)、转基因大豆(Glycine max)GTS40-3-2、转基因玉米(Zea mays)Bt11等标准物质购于欧盟联合研究中心标准物质与测量研究所及美国油脂化学家协会。各种含量的转基因克螟稻、TT51-1、KF6等转基因大米标准样品及非转基因大米(明恢63)、大豆、玉米、小麦,高粱,油菜等样品由本实验室保存。
ddPCR Super Mix(no dUTP)、ddPCR Droplet Generation Oil、ddPCR Droplet Reader Oil、Droplet Generator DG8 Cartridge 美国Bio-Rad公司;RealMaster Mix(with ROX) 美国ABI公司;食品基因组提取试剂盒NucleoSpin®Food 美国MN公司。
1.2 仪器与设备
QX200™微滴式数字PCR系统(包括微滴生成器、封膜机、PCR扩增仪和微滴读取仪) 美国Bio-Rad公司;Bio-Mark™微流体芯片式数字PCR系统(含仪器配套耗材) 美国Fluidigm公司;QuantStudio™ 3D微流体芯片式数字PCR系统(含仪器配套耗材) 美国Life Technologies公司;7500实时荧光PCR仪 美国ABI公司;样品研磨机 德国IKA公司;离心机 美国Beckman Coulter公司;Qubit核酸蛋白分析仪 美国Thermo Scientific公司。
1.3 方法
1.3.1 基因组DNA提取
所有样品材料放入样品研磨机中,研磨至粒度60 目左右,样品基因组DNA按照NucleoSpin®Food核酸提取试剂盒说明书提取,-20 ℃保存备用。
1.3.2 引物和探针的设计及合成
本实验根据大米蔗糖磷酸合成酶(sucrose phosphate synthase,SPS)和转基因克螟稻品系特异性基因序列,分别设计3 对引物探针(表1),用于筛选适用于数字PCR的引物探针组,相关序列及其标记由英潍捷基(上海)贸易有限公司合成。
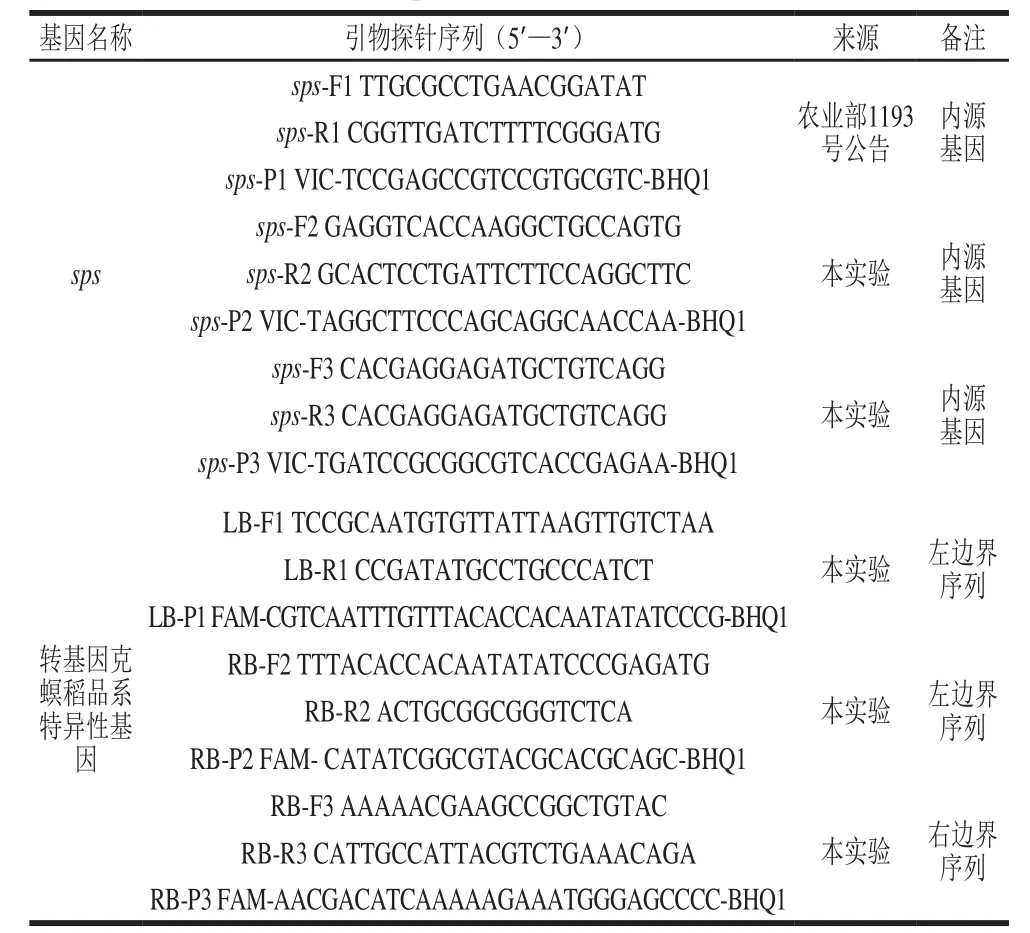
表 1 sps和克螟稻品系特异性基因引物探针序列Table 1 Sequences of primers and probes for used for PCR amplification of sps and KMD event-specific genes
1.3.3 扩增体系和反应参数
1.3.3.1 数字PCR扩增体系
分别向0.2 mL PCR管中加入2×ddPCR Super Mix(no dUTP)10 μL,终浓度为0.32 µmol/L的sps基因及克螟稻品系特异性基因正向引物和反向引物,0.12 µmol/L sps基因荧光标记探针,0.2 µmol/L克螟稻品系特异性基因荧光标记探针,1 µL DNA模板,利用ddH2O补齐至20 µL。
1.3.3.2 数字PCR扩增参数及数据分析
将配制好的数字PCR混合液,加入微反应体系生成装置的加样孔中生成微反应体系。将微反应体系放置于PCR扩增仪中,按以下参数进行PCR扩增:95 ℃、5 min(1 ℃/s),1 个循环;94 ℃、15 s,60 ℃、1 min(1 ℃/s),50 个循环;98 ℃、10 min(1 ℃/s),1 个循环;12 ℃保存反应产物。将扩增产物放入读取仪中对微反应体系进行荧光检测并计数。
1.3.3.3 实时荧光PCR扩增体系和参数
0.2 mL PCR反应管中加入2×Real Master Mix(with ROX)10 μL,终浓度为0.4 µmol/L的正向引物和反向引物、0.2 µmol/L的探针、1 µL DNA模板,利用ddH2O补齐至20 µL。
实时荧光PCR程序为:预变性95 ℃、10 min;95 ℃、10 s;60 ℃、45 s;45 个循环。
1.3.4 引物探针的筛选及其特异性
以常见转基因作物品系和常见植物基因组DNA为模板,按上述反应条件对本研究各组引物探针的有效性和特异性进行测试。分别采用非转基因大米明恢63和转基因克螟稻品系大米样品DNA为扩增模板,以ddH2O为空白对照,分别对3 组sps基因和克螟稻品系特异性基因的引物探针进行有效性扩增。随后选取扩增效率最高的sps基因和克螟稻品系特异性基因引物探针组,利用非转基因大米明恢63、转基因大米TT51-1、KF6 、LL62品系、大豆、玉米、小麦、高粱、油菜、100%及1%转基因克螟稻大米样品、转基因大豆GTS40-3-2、转基因玉米Bt11为模板,验证扩增效率最高的sps基因和克螟稻品系特异性基因引物探针组的特异性。
1.3.5 转基因克螟稻定量体系的条件优化
利用Qubit核酸蛋白分析仪将质量分数为100%的转基因克螟稻DNA溶液定量到质量浓度20 ng/µL,并按6 倍梯度稀释后分别加入4 µL DNA溶液至内源基因sps的微滴式数字PCR扩增体系中,每个梯度3 次重复进行定量实验,通过计算每个梯度3 次重复的相对标准偏差(relative standard deviation,RSD),判断转基因克螟稻定量体系中最适DNA模板添加量。
以质量分数为100%的转基因克螟稻大米样品为模板,分别考察扩增退火温度和单双重微滴式数字PCR等扩增条件对定量结果的影响。将微滴式数字PCR扩增参数中的退火温度分别设为58、60、62 ℃和64 ℃,每个梯度3 次重复进行定量实验,按1.3.3节所述方法计算最终的克螟稻转基因成分含量。根据优化好的条件,分别比较sps基因和克螟稻品系特异性基因分开扩增的两个单重微滴式数字PCR体系与两个基因合并后的双重微滴式数字PCR体系扩增5 次重复结果的差异。
1.3.6 转基因克螟稻定量方法在芯片式数字PCR平台上的适用性
分别在微流体芯片式数字PCR平台Bio-Mark™和QuantStudio™ 3D对已建立的基于微滴式数字PCR平台的转基因克螟稻成分定量检测方法进行验证。利用已优化的双重数字PCR条件,将质量分数为100%的转基因克螟稻标准样品分别在Bio-Mark™和QuantStudio™ 3D微流体芯片式数字PCR平台上进行扩增,实时监测其扩增曲线,以确保每个微滴/微孔均能有效扩增,针对扩增微反应体系的分布情况等逐一进行分析。
1.3.7 转基因克螟稻定量的绝对灵敏度和线性范围
利用Qubit核酸蛋白分析仪测定转基因克螟稻大米DNA溶液浓度后按2 倍梯度稀释,分别稀释至1.5 pg及0.5 pg,即内外源基因的浓度均为1 copies/μL(大米单拷贝基因组质量约为0.5 pg[25])。共稀释4 个梯度,每个梯度3 个重复实验,按1.3.3节进行微滴式数字PCR扩增,分别检测其内源基因sps和克螟稻品系特异性基因的拷贝数,根据内外源基因的理论值和实际值绘制相关性曲线。
1.3.8 数字PCR与实时荧光PCR对不同含量转基因克螟稻的测定
利用上述实验建立的双重数字PCR定量体系,分别在QX200™微滴式数字PCR平台和QuantStudio™ 3D微流体芯片式数字PCR平台上对质量分数为0.1%、1%、5%的转基因克螟稻样品的进行定量分析,每个样品3 次重复定量实验,根据仪器自动给出的内外源基因拷贝数浓度,计算二者之比即为样品中的转基因克螟稻含量。
根据1.3.3节的实时荧光PCR扩增体系和参数,利用已知质量分数为100%、10%、5%、0.5%及0.1%的转基因克螟稻样品进行实时荧光PCR扩增,基于扩增Ct值制作内外源基因的标准曲线,并将0.1%、1%、5%转基因克螟稻样品内外源基因Ct值代入标准曲线中,对这3 个含量的样品进行定量,将其结果与微滴式及芯片式数字PCR定量结果比较研究。
1.4 数据分析
数字PCR扩增结束后,每个基因荧光本底值与阳性值之间差异较大,即阴阳性扩增热点之间界限分明且阳性扩增热点较为集中,即可判定为扩增结果良好,扩增图像可直接用作实验结果。
由于数字PCR是绝对定量技术,无需标准曲线即可直接测得样中的内外源基因绝对含量。因此,数字PCR读取仪逐一对每个微反应扩增产物进行计数分析后,根据泊松分布原理计算得到sps和克螟稻品系特异性基因的拷贝数浓度(copies/μL)。而本研究采用的sps和克螟稻品系特异性基因均为单拷贝基因,按下式计算样品中转基因克螟稻定量结果[26-27]:

2 结果与分析
2.1 引物探针筛选
通过采用实时荧光PCR方法对已设计合成的内外源基因引物探针进行筛选,结果表明所有引物探针均能有效扩增出靶基因,但sps基因引物探针组F2/R2/P2和克螟稻品系特异性基因引物探针组LB-F1/R1/P1信号最强(图1)。为保证后续数字PCR能到达最大效率,本研究选取sps-F2/R2/P2和LB-F1/R1/P1两组引物探针,用于转基因克螟稻的内外源基因定量。
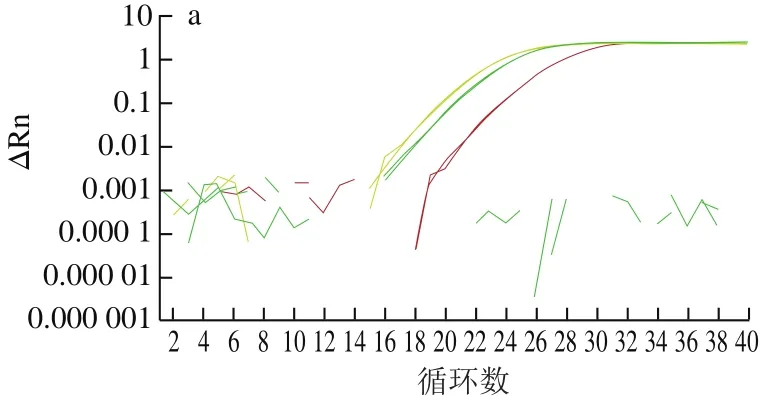

图 1 sps及克螟稻品系特异性基因的引物探针筛选结果Fig. 1 Effectiveness of the primers and probes in amplifying sps and KMD event-specific genes

图 2 sps- F2/R2/P2及LB-F1/R1/P1引物探针组特异性扩增结果Fig. 2 Specificity of the primers and probes in amplifying sps and KMD event-specific genes
以利用非转基因大米、常见转基因大米、大豆及玉米品系及常见作物基因组为模板,对sps-F2/R2/P2和LB-F1/R1/P1两组引物探针的特异性进行分析。sps-F2/R2/P2引物探针组仅能扩增出大米成分(含非转基因大米明恢63、转基因大米TT51-1、KF6、LL62品系),对其他禾本科植物玉米、小麦、高粱及大豆、油菜等作物均无扩增。LB-F1/R1/P1引物探针组仅能扩增出克螟稻转基因成分,对其他转基因大米品系和其他转基因作物均无扩增(图2)。表明这两组引物探针特异性较好。
2.2 转基因克螟稻微滴式数字PCR定量体系的条件优化
当加入数字PCR扩增体系中的克螟稻大米样品DNA量从20 ng/µL按6 倍稀释6 个梯度后,20 µL扩增体系中DNA量在10 pg~80 ng之间时,目标基因的理论拷贝数浓度约为8 000、1 333、222、37、6、1 copies/µL。当其含量高达8 000 copies/µL即扩增体系中DNA浓度高达80 ng时,6 次重复的RSD高于25%,而其余几个浓度梯度,即使DNA含量低至1 copies/µL,6 次重复的RSD仍低于25%(表2),即本研究中克螟稻定量体系中最适DNA添加量在10 pg~13 ng之间。
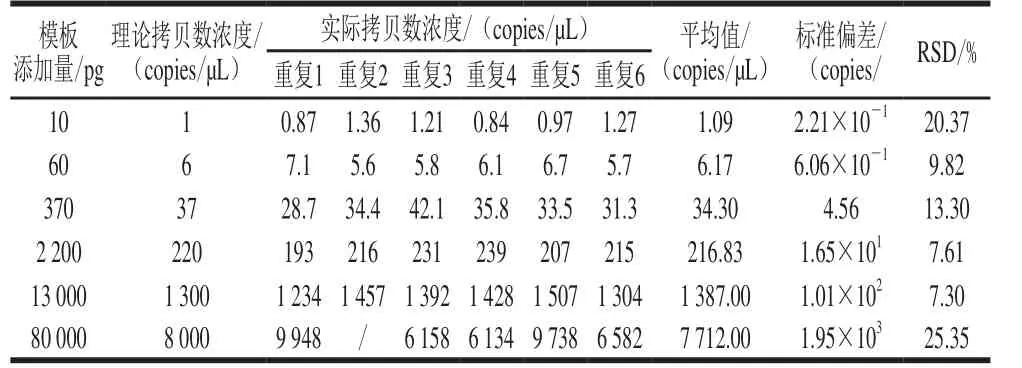
表 2 克螟稻定量体系模板优化Table 2 Optimization of genomic DNA concentration in the quantitative system
当数字PCR扩增退火温度分别为58、60、62 ℃和64 ℃时,质量分数为100%的克螟稻转基因相对含量分别为99.29%、100.36%、100.43%、97.39%,所有扩增样品均能有效区分阴性和阳性微滴点(图3)。虽然当退火温度为60 ℃时偏差最小,但统计学分析显示这4 组数据无显著性差异(P>0.05),即退火温度在58~64 ℃之间时,对定量结果无显著性影响。

图 3 克螟稻定量体系PCR退火温度优化Fig. 3 Optimization of annealing temperature for GM rice KMD in digital PCR
以质量分数为100%的转基因克螟稻大米样品为模板,根据优化条件,在模板含量为10 ng DNA,退火温度为60 ℃时,比较分开扩增sps基因和克螟稻品系特异性基因的两个单重数字PCR体系与两个基因合并后的双重数字PCR扩增体系定量结果。单重扩增后定量得到的克螟稻转基因相对含量均值为100.11%,RSD为1.74%(图4a),而双重体系的相对含量均值为99.93%,RSD为3.59%(图4b)。两种方法定量偏差及多次重复的RSD均远小于25%,符合目前国际最严格的国际标准和欧盟定量方法要求[28-29],即两种方法均适用于克螟稻大米样品的定量。为节约试剂和时间成本,后续实验中,均采用双重数字PCR扩增体系进行研究。

图 4 单重数字PCR与双重数字PCR扩增结果比较Fig. 4 Comparison of amplification results between singplex and duplex digital PCR
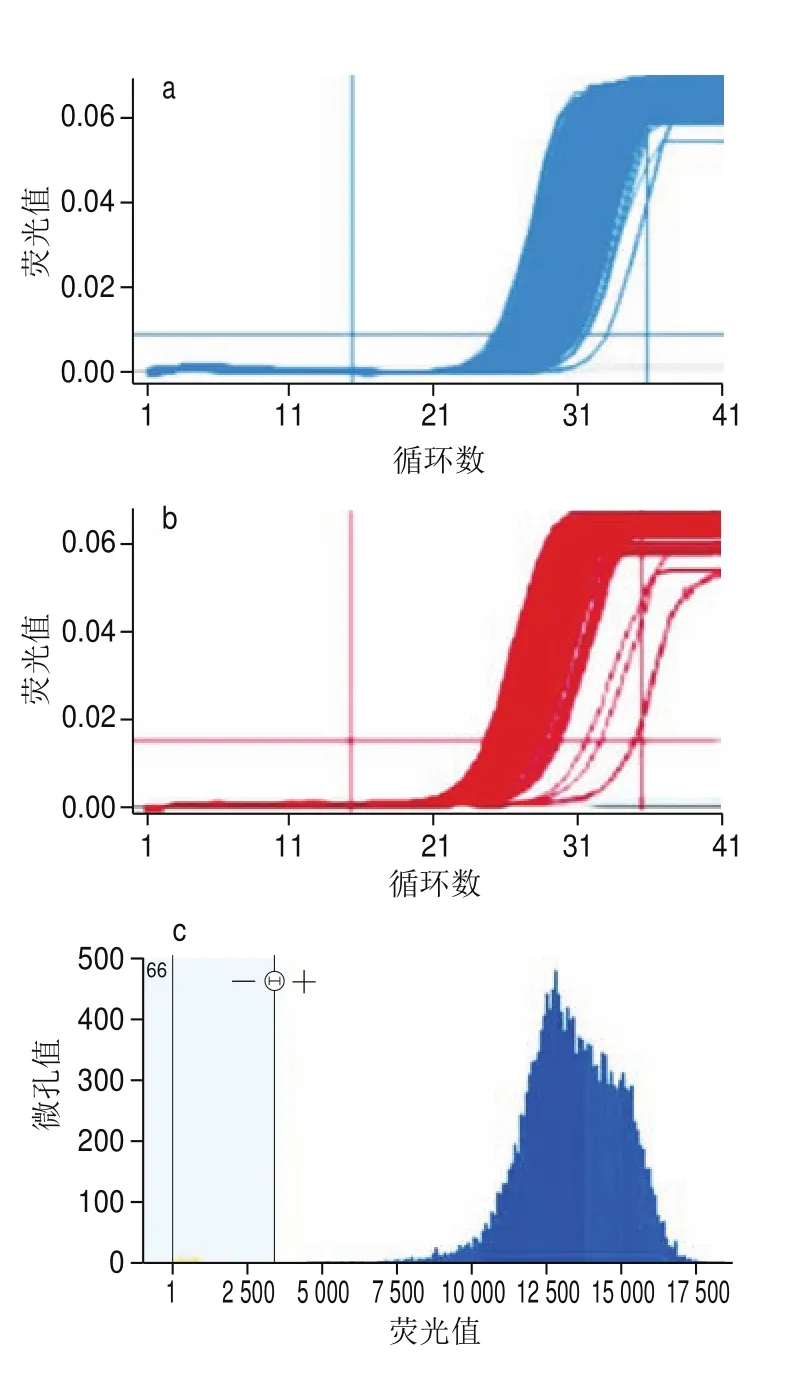
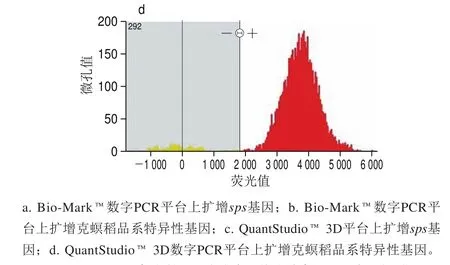
图 5 100%克螟稻转基因大米在芯片式数字PCR平台上扩增Fig. 5 Amplification of KMD GM rice genes on the chip-based digital PCR platform
转基因克螟稻定量方法在芯片式数字PCR平台上的适用性利用已优化的微滴式数字PCR扩增条件,分别将质量分数为100%的克螟稻大米样品在微流体芯片式数字PCR平台Bio-Mark™和QuantStudio™ 3D上进行扩增,分别得到其内源基因与外源基因的实时扩增曲线(图5)。Bio-Mark™数字PCR平台上内外源基因各自的765 个反应仓Ct值均较为集中,表明所有微孔均得到了较好的扩增,这也使得定量结果准确可靠(图5a、b),相对含量为103.26%,3 次重复的RSD仅为1.7%,定量准确性和精密度均较为理想。QuantStudio™ 3D微流体芯片式数字PCR平台对质量分数为100%的标准样品进行芯片式数字PCR定量检测时,内外源基因所产生的阴性点和阳性点分离较好,无中间模糊区(图5c、d),相对含量为99.43%,3 次重复的RSD仅为1.7%。表明本研究所建立的微滴式数字PCR定量方法在其他微流控芯片式数字PCR平台上也能正常扩增,本方法适用性较好。
2.3 转基因克螟稻定量绝对灵敏度和线性范围
将克螟稻转基因大米DNA溶液按多倍梯度稀释时,在1~1 000 copies/μL的整个稀释梯度中,sps与克螟稻品系特异性基因RSD分别在0.63%~2.62%和0.96%~3.62%之间,所有稀释梯度的RSD均符合国际要求[29]。其内外源基因稀释梯度理论值和实际值线性相关,R2分别为1和0.999 8。该体系所检测的浓度梯度均具有很好的线性关系,并且与理论拷贝数相符,该检测体系能够对含量低至1 copies/μL的转基因克螟稻进行准确定量。
2.4 转基因克螟稻数字PCR定量结果与实时荧光PCR方法定量结果比较
运用已建立的数字PCR方法,分别在QX200™微滴式数字PCR平台、QuantStudio™ 3D微流体芯片式数字PCR平台及实时荧光PCR平台上定量分析转基因质量分数为0.1%、1%、5%的克螟稻转基因大米样品,将三者结果进行比较分析。所有平台定量结果的3 次重复RSD均小于25%,表明所有操作重复性较好。微滴式和芯片式数字PCR平台上3 个梯度转基因大米样品定量偏差在-0.204%~7.203%之间(表3),即使是针对质量分数为0.1%的克螟稻转基因大米样品,该方法在不同平台上的定量结果精密度和准确性仍然符合要求。表明本研究建立的数字PCR方法对克螟稻的定量检测灵敏度可达0.1%。
利用实时荧光PCR方法对质量分数同样为0.1%、1%、5%克螟稻样品进行定量时,内外源基因的实时荧光PCR扩增效率分别为107.34%和99.62%,符合90%~110%的范围[30]且内外源基因的标准曲线R2都达到0.99以上。根据各组数据RSD可知,本研究中实时荧光PCR的实验操作重复性较好,且内外源基因PCR扩增效率及标准曲线相关性也符合要求,而3 个梯度转基因大米样品的定量值其与实际值偏差分别为29.942%、25.426%及-6.738%(表3),远大于微滴式及芯片式数字PCR的偏差,尤其是0.1%和1%的克螟稻样品定量偏差均已超过目前国际通用的定量方法要求[29]。而数字PCR即使针对质量分数为0.1%的样品,其偏差最大也仅为7.203%,符合转基因定量中偏差在25%以内的要求[28-29]。因此,数字PCR的定量准确性及灵敏度要好于实时荧光PCR,尤其是针对低含量的克螟稻成分进行定量时,数字PCR的结果更为可靠。
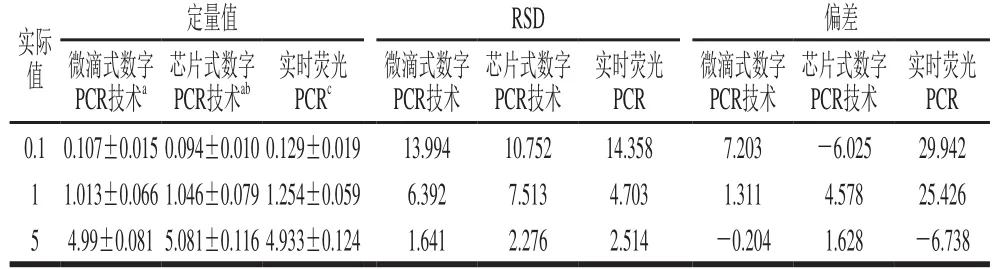
表 3 不同含量的克螟稻数字PCR及实时荧光PCR定量检测结果Table 3 Quantitative results of different contents of KMD GM rice by digital PCR and real-time PCR%
3 讨论与结论
虽然数字PCR技术将DNA模板进行稀释分布到大量的独立反应室中进行单独扩增,极大降低了反应间的相互抑制,提高了反应效率,但也有研究表明DNA纯化方法等条件优化措施会对定量结果产生较大的影响[24,31]。因此本研究对最能影响克螟稻定量反应体系的因素如模板浓度、退火温度及单双重反应的差异等情况均进行研究,建立适用于克螟稻品系特异性转基因成分定量的最优条件。还有研究表明模板DNA的片段长度会影响模板的均匀分布,进而影响定量结果[11],本研究分析随机酶切、超声等方式断裂大米基因组DNA对数字PCR定量结果的影响,发现当DNA浓度在合适范围内时,各种处理后的定量结果与对照相比,其差别无统计学意义(数据未列出),即大米基因组DNA无需断裂处理也可得到较为理想的定量结果。但此结果仅适用于克螟稻等转基因大米品系的定量研究,对于其他转基因作物,尤其是小麦等基因组较大的作物,在对样品精准定量前还需对数字PCR的各种条件逐一进行优化。
在转基因定量中考虑测量值的不确定度很有必要,由于对一些影响因素缺少定量描述,因此在转基因成分定量检测的不确定度分析方面还没有一个通行的成熟方法,欧盟联合研究中心推荐采用循环实验结果进行某一转基因定量方法的不确定度评估[32],在缺乏循环实验数据时,可以利用现有数据依据国家计量技术规范[33]和文献[30,34]方法对定量的不确定度进行分析。由于数字PCR无需标准曲线对样品进行定量,由标准物质均匀性、稳定性、稀释梯度及标准曲线等因素带来的不确定度均无需考虑,这也是利用实时荧光PCR定量检测转基因成分时最能影响不确定度的部分[35]。此外,数字PCR为终点读取,PCR扩增效率的不确定度理论上也可排除在外。因此除取样的不确定度之外,仅需考虑稀释梯度、批内重复、仪器示值等的不确定度即可。
转基因含量通常以质量、体积或者单位个数(颗粒、DNA以及蛋白分子等)的形式描述纯的转基因成分的质量与相应成分的总质量之比。数字PCR是通过绝对定量方法测定目的基因在样本中的拷贝数,通过内外源基因的拷贝数之比得到转基因成分的含量。在目标基因为单拷贝插入的转基因品系中,所得内外源基因拷贝数之比即为转基因含量百分比。若目标基因为多拷贝插入或样本为亲本杂交后代,该比值作为相对定量结果将不够准确,需作进一步换算。对于定量中的内源基因,首先要选择低拷贝且拷贝数可稳定遗传的基因,由于单拷贝基因相对于多拷贝基因来说更加稳定,在不同作物栽培种中的突变率更低[36],更加适合用作转基因成分定量中的内源基因。而转基因定量分析中的外源基因选择时,单拷贝的特异边界序列是首选,尤其是当检测样品为混合产品时,对每一品系分别进行定量,得到的转基因成分含量比利用某一外源筛选元件进行定量得到的结果要更为准确。但也有研究指出以多拷贝基因为对象的检测由于原始材料中基因的拷贝数高使得检测灵敏度更高[37]。本研究中大米内源基因sps和克螟稻品系特异性基因均为稳定遗传的单拷贝基因,因此内外源基因拷贝数之比可直接转换为克螟稻转基因成分含量。
转基因标识制度的有效实施依赖于科学可靠的转基因定量检测方法。本研究结果表明,基于数字PCR的定量方法可准确检测样品中的从0.1%~100%质量分数的克螟稻转基因大米成分,尤其是在对低含量克螟稻转基因大米成分的定量中,方法的精密度和准确性均高于目前常用的实时荧光PCR方法,可以满足转基因标识需求,也为我国转基因产品标识阈值的设定提供了一定的借鉴依据。
猜你喜欢
贵州畜牧兽医(2023年3期)2023-06-29 07:07:28
农技服务(2023年2期)2023-03-15 00:43:08
学与玩(2022年10期)2022-11-23 08:32:00
今日农业(2022年3期)2022-06-05 07:12:08
园林科技(2020年2期)2020-01-18 03:28:18
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
创新科技(2015年1期)2015-12-24 06:23:21
物理实验(2015年9期)2015-02-28 17:36:47
作物研究(2014年6期)2014-03-01 03:39:03
