
HPLC法测定富马酸沃诺拉赞中富马酸含量方法研究
2019-04-27 07:39:36张羽强
山东化工 2019年7期
张羽强
(海南南陆顺捷食品药品检测有限公司,海南 海口 570311)
富马酸沃诺拉赞,化学名为5-(2-氟苯基)-N-甲基-1-(3-吡啶基磺酰基)-1H-吡咯-3-甲胺富马酸盐,是日本武田制药(Takeda) 研发的钾离子竞争性酸阻滞药,于2014年12 月首次在日本上市,对糜烂性食管炎、幽门螺杆菌感染、十二指肠溃疡、胃溃疡等胃酸相关性疾病 (ARDs) 疗效良好[1-4]。
富马酸含量测定在原料药质量控制中作为重要的控制参数之一,需要建立含量的测定方法,参照国内外药典,均未收载该品种的原料药质量标准,因此,通过进行本品富马酸含量测定方法的摸索及方法验证,对其药品质量控制提供了参考依据。
1 材料与方法
1.1 仪器
LC2030C (日本岛津)、CPA225D型十万分之一天平、FA2014N型万分之一天平、PB-10酸度计。
1.2 试药
富马酸沃诺拉赞原料(批号:S00015、S00023、S00029),富马酸对照品(中国食品药品检定研究院,批号:111541-201102),富马酸沃诺拉赞对照品(自制);乙腈为色谱纯,磷酸、磷酸钠均为分析纯,水为纯化水。
2 方法与结果
2.1 色谱条件
色谱柱为十八烷基硅烷键合硅胶为填充剂(Waters Symmetry C18 柱(4.6 mm×250 mm,5 μm)的色谱柱,流动相:0.02 mol/L磷酸钠溶液(取磷酸钠6.4 g,置1000 mL量瓶中,加水溶解并稀释至刻度,摇匀,用磷酸调节pH值至7.0)-乙腈(80∶20);检测波长:230 nm,流速:1.0 mL/min,进样量:10 μL。
2.2 溶液的配置
(1)对照品溶液 精密称取富马酸沃诺拉赞对照品适量,加流动相超声溶解并稀释制成每1 mL约含0.2 mg的溶液。(2)富马酸对照溶液:精密称取富马酸对照品适量,加流动相溶解并稀释制成每1 mL约含50 μg的溶液。(3)供试品溶液 取供试品约20 mg,精密称定,置于100 mL量瓶中,加溶剂溶解并稀释至刻度,摇匀,即得。
2.3 方法学验证
2.3.1 系统适用性
精密量取对照品溶液10 μL注入高效液相色谱仪中,记录色谱图,结果见表1。

表1 系统适用性试验结果
2.3.2 进样精密度试验
精密量取对照品溶液10 μL注入高效液相色谱仪中,连续进样6次,记录色谱图,结果见表2。

表2 进样精密度试验结果
2.3.3 专属性试验
(1)氧化破坏试验:精密称取供试品约10 mg,置50 mL量瓶中,加3%的过氧化氢溶液 2 mL,室温破坏20 h,用溶剂超声溶解并稀释至刻度,摇匀,即得。(2)强酸破坏试验:精密称取供试品约10 mg,置50 mL量瓶中,加2 mol/L盐酸溶液2 mL,破坏10 h,加2 mol/L氢氧化钠溶液2 mL中和,摇匀,加溶剂稀释至刻度,摇匀,即得。(3)强碱破坏试验:精密称取供试品约10 mg,置50 mL棕色量瓶中,加2 mol/L氢氧化钠溶液2 mL,破坏10 h,用2 mol/L盐酸溶液2 mL中和,加溶剂稀释至刻度,摇匀,即得。(4)高温破坏试验:精密称取供试品约10 mg,置50 mL量瓶中,加溶剂适量,于水浴破坏5 h,放冷至室温,用溶剂稀释至刻度,摇匀,即得。(5)光照破坏试验:精密称取供试品约10 mg,置50 mL无色量瓶中,加溶剂超声溶解并稀释至刻度,在光照强度为4500lx±500lx的培养箱照射10 h。
精密量取上述溶液各10 μL,分别注入液相色谱仪,记录色谱图,试验结果可知,在强酸、强碱、氧化、高温、光照破坏条件下,空白溶剂无干扰,富马酸纯度指数值均大于0.99,本法专属性良好能满足含量测定要求。
2.3.4 线性与范围
照2.2项下溶液配制方法,配制线性系列溶液。分别精密量取10 μL注入液相色谱仪,记录色谱图。试验结果见表3、图1。
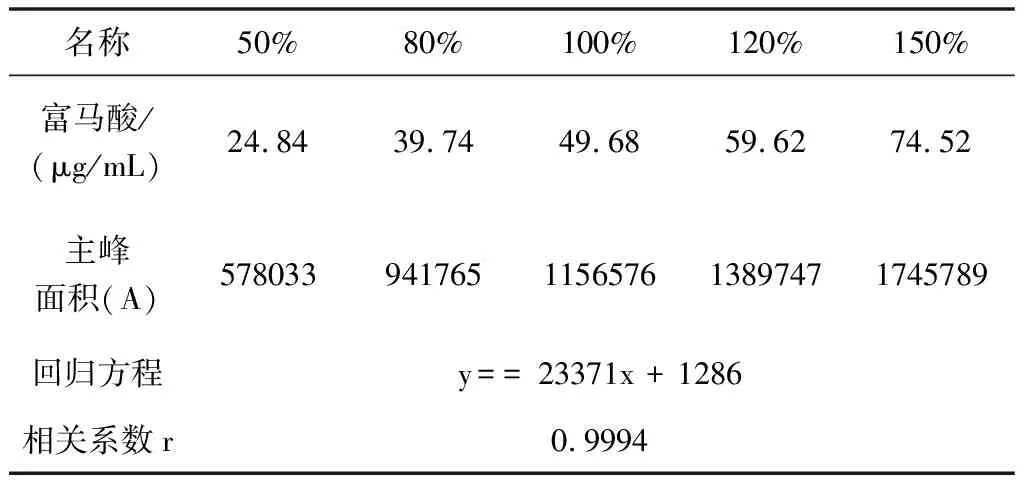
表3 富马酸线性关系试验结果
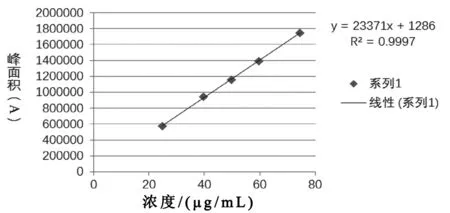
图1 富马酸线性关系图
2.3.5 供试品精密度试验
2.3.5.1 重复性试验
取本品,称取6份,照2.2项下溶液配置,分别精密量取10 μL注入液相色谱仪,记录色谱图。试验结果见表4。
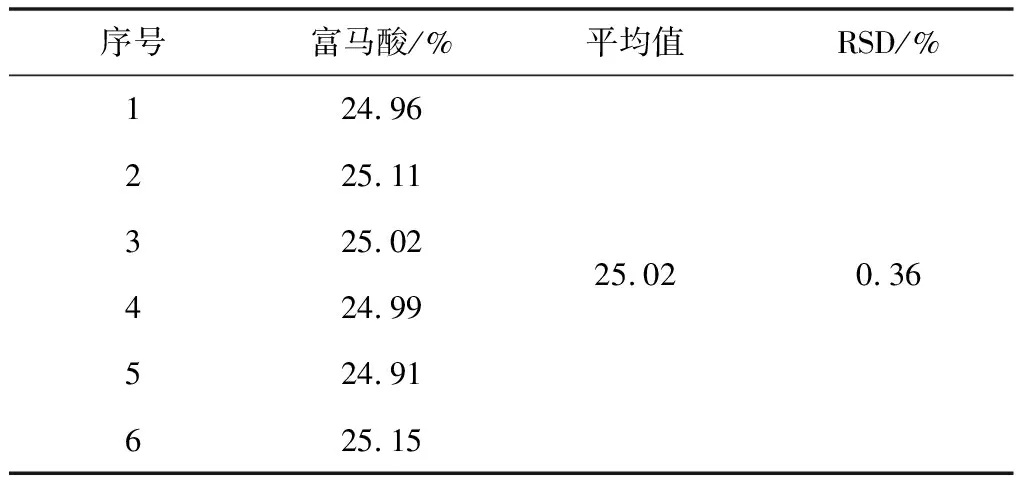
表4 重复性试验结果
2.3.5.2 中间精密度
参照重复性试验的测定方法,在相同的试验室由不同人员、不同的仪器及不同的时间进行试验。试验结果见表5、6。
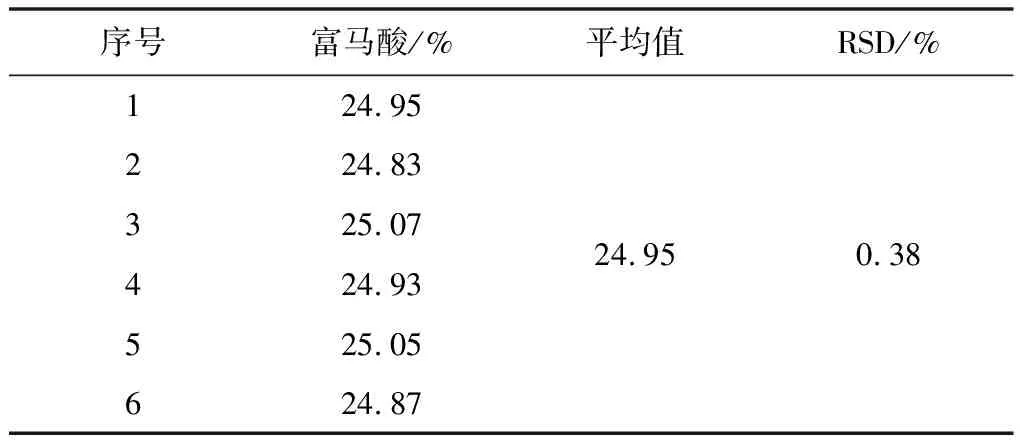
表5 中间精密度试验结果(n=6)
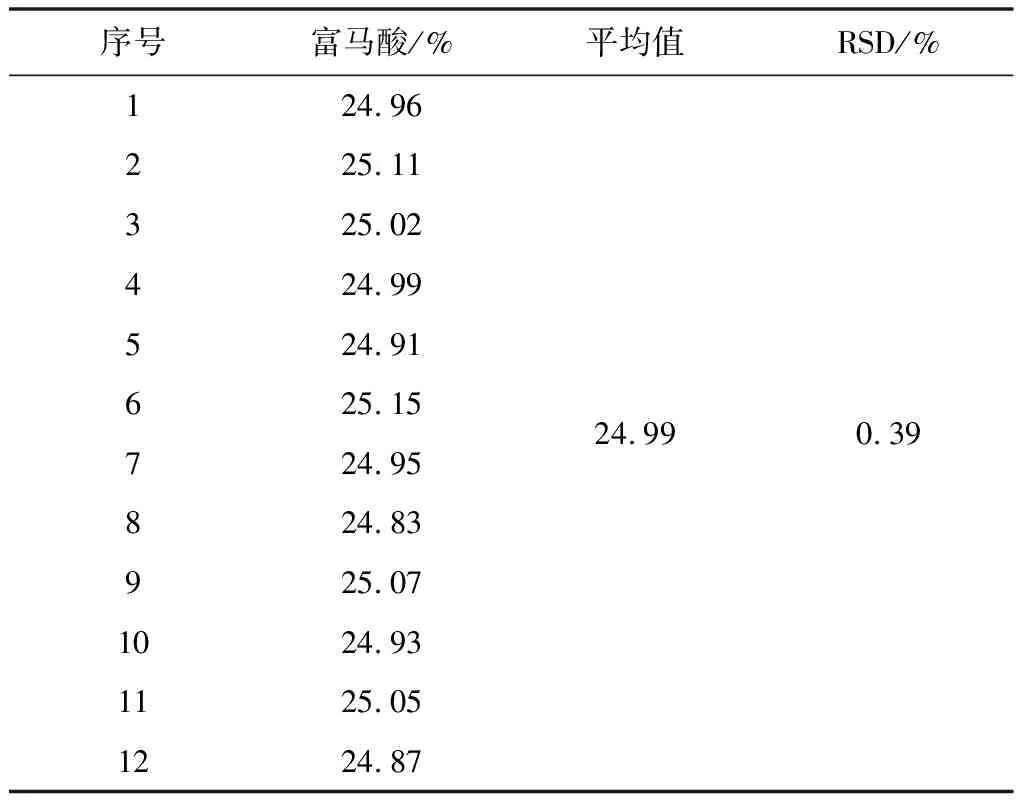
表6 中间精密度试验结果(n=12)
2.3.6 溶液稳定性
取同一份样品溶液,置室温下放置,分别于0、2、4、6、8、10 h取样,精密量取10 μL注入液相色谱仪,记录色谱图。试验结果见表7。
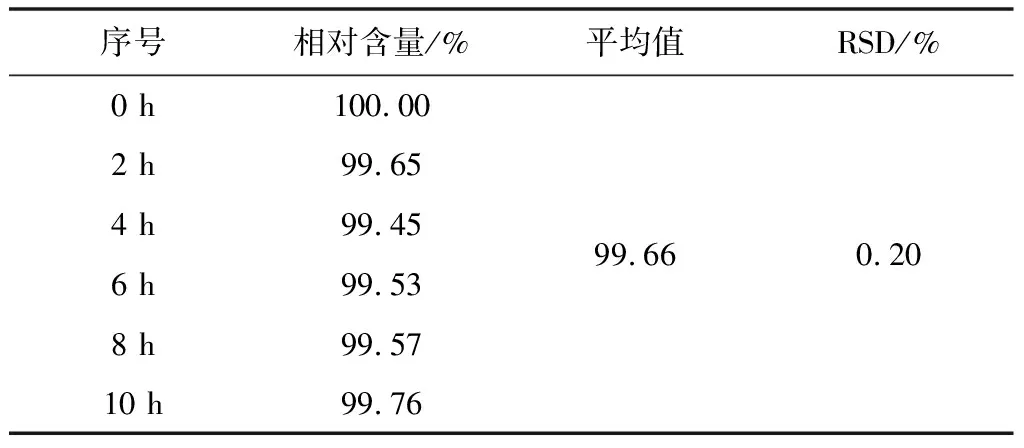
表7 溶液稳定性试验结果
2.3.7 准确度试验
照2.2项下配制方法,分别配制80%、100%、120%溶液各3份,精密量取10 μL注入液相色谱仪,记录色谱图。试验结果见8。
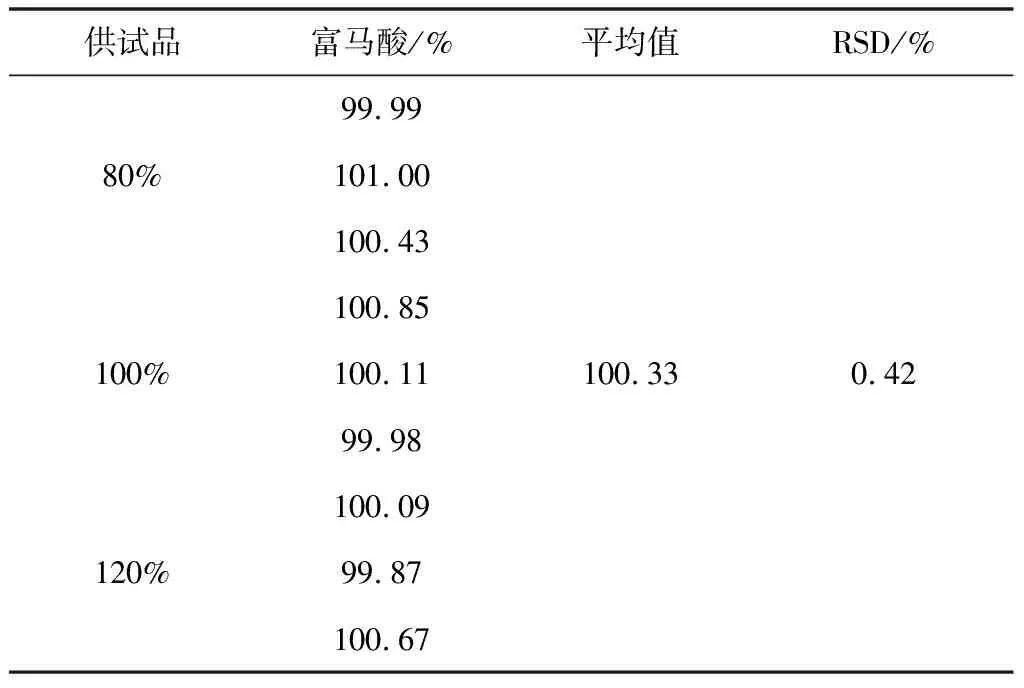
表8 富马酸回收率试验结果
2.3.8 耐用性试验
照2.2项下方法配制样品,根据液相色谱法中典型的变动因素进行试验。其变动因素如下:流速、流动相的组成、流动相pH值、色谱柱的改变进行耐用性试验考察。试验结果说明,改变以上变动因素均能满足本品富马酸及富马酸沃诺拉赞含量检测要求。试验结果见表9。
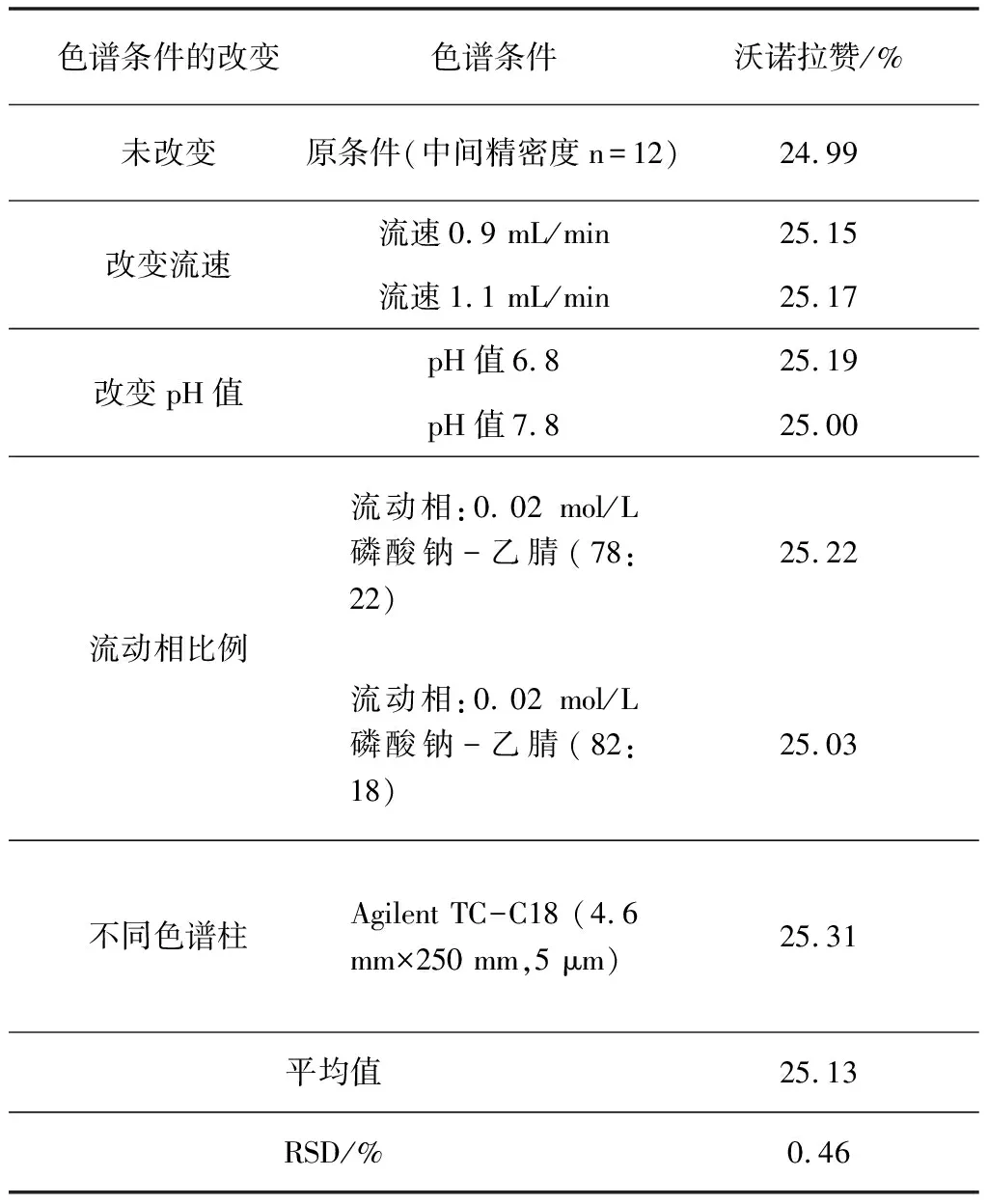
表9 耐用性供试品含量变化结果
2.3.9 样品测定
取三批富马酸沃诺拉赞,按本法进行含量检测,结果分别为 25.02%、24.98%、25.09%。
3 讨论
(1)根据波长扫描,富马酸的吸收最大波长为203.60 nm。由于本品有最大吸收紫外末端,根据二极管阵列检测器进行波长选择,波长为230 nm富马酸色谱峰形最佳,溶剂空白无干扰,故选择230 nm作为本品富马酸的测定波长。
(2)在流动相筛选过程中,采用0.02 mol/L磷酸盐缓冲液(用磷酸调节pH值至7.0) -乙腈(V∶V= 70∶30)、0.02 mol/L磷酸盐缓冲液(用磷酸调节pH值至7.0) -乙腈(V∶V=80∶20)等条件,在采用0.02 mol/L磷酸盐缓冲液(用磷酸调节pH值至7.0) -乙腈(V∶V= 70∶30) 为流动相时,富马酸色谱峰保留时间及峰形较佳,重复性最好,空白溶剂无干扰,富马酸峰纯度也能满足含量测定要求。
4 结论
通过对富马酸测定方法详细的验证,验证结果表明,该方法专属性良好、准确、快捷,精密度及耐用性好;可用于富马酸沃诺拉赞中富马酸的含量测定,并对其药品质量控制提供了参考依据
猜你喜欢
食品安全导刊(2021年21期)2021-08-30 08:21:40
皮肤病与性病(2021年3期)2021-07-30 08:08:48
皮肤病与性病(2021年3期)2021-07-30 08:08:02
科学与财富(2021年35期)2021-05-10 11:54:37
食品安全导刊(2020年14期)2020-12-04 20:19:39
食品安全导刊·中旬刊(2020年5期)2020-06-04 08:43:53
哲思(2017年8期)2017-11-01 11:56:56
知识窗(2017年7期)2017-07-31 11:34:10
时代青年(上半月)(2017年7期)2017-07-24 08:47:44
化工技术与开发(2013年1期)2013-09-27 06:43:28
