
Antidiabetic treatment on memory and spatial learning:From the pancreas to the neuron
2019-04-16 02:21:54EleniXourgiaAthanasiaPapazafiropoulouAndreasMelidonis
World Journal of Diabetes 2019年3期
Eleni Xourgia,Athanasia Papazafiropoulou,Andreas Melidonis
Eleni Xourgia,Athanasia Papazafiropoulou,Andreas Melidonis 1st Department of Internal Medicine and Diabetes Center,Tzaneio General Hospital of Piraeus,Athens 18536,Greece
Abstract
Key words:Memory;Spatial learning;Cognitive;Neural remodeling;Type 2 diabetes mellitus;Antidiabetic drugs
Diabetic encephalopathy (DE) is defined as a complex combination of central nervous system (CNS) structural and functional changes,stemming mostly from oxidative stress and chronic inflammation of the neural tissue in the setting of long-standing hyperglycemia.While several mechanisms have been proposed for the explanation of cognitive decline in diabetic subjects,the intricate interplay of various signaling pathways along with the numerous co-morbidities of patients with diabetes do not allow for a definite pathogenetic model to be proposed.Moreover,the pathophysiological substrate of type 2 diabetes mellitus (T2DM) encephalopathy appears to be different from that of T1DM DE[1].Currently,despite the abundance of evidence of the subject,the molecular mechanisms implicated in the development of DE and its rate of progression have not been clarified,resulting in a subsequent lack of treatment options for interruption or reversal of the cumulative neuronal damage and functional decline of patients.The purpose of our review is to summarize and describe the interaction between the various antidiabetic substances and DE,in order to facilitate the possible development of a therapeutic algorithm for affected patients.
ENCEPHALOPATHY IN T2DM
Several studies in T2DM subjects have confirmed the dysfunction of cognitive capacity,both in executive and processing tasks,when compared to healthy controls[2-5].While the decline of neural capacity in T2DM is described as a multifactorial process,it is evident that tissue insulin resistance (IR) plays a pivotal role in the pathogenetic process.Insulin receptors are expressed in all major components of the CNS (neurons,microglia,astrocytes,oligodendrocytes and vascular system) in varying degrees.The downstream effects of insulin signaling in neural tissue include neurogenesis,apoptosis inhibition,cytokine release,attenuation of inflammatory response,vasodilation and glucogen uptake and storage[6].While some researchers have proposed the possibility of de novo insulin synthesis in the CNS,current experimental data support the fact that the majority of centrally-acting hormone is produced at the pancreatic β-cells and subsequently transported through the blood brain barrierviathe systemic circulation,with vascular endothelium significantly affecting the process[7].The role of other peripherally-acting hormones such as glucagon-like peptide-1 (GLP-1),leptin or ghrelin on insulin transport and potency in the CNS has not been described so far.IR,defined as a dysfunction on any of the several stages preceding or during the signaling cascade activated by the insulinreceptor complex formation,can affect the homeostasis of all the processes described above that are mediated by the hormone.
ANTIDIABETIC TREATMENT AND NEURAL FUNCTION
Biguanides
The information surrounding metformin and its effect on cognitive impairment is contradictory and highly complex,varying between different types of test subjects and changing in accordance to different treatment dosages and pathophysiological substrates studied.On a cellular level,metformin exhibits pleiotropic effects,including interaction with multiple signaling pathways such as those of mitogenactivated protein kinases (MAPK) and mammalian target of rapamycin complex 1,that are closely linked to proliferation and apoptosis.Given the relative safety of the substance and its role in cellular turnover,the possibility of repurposing it for use in neurofunctional disorders is currently being investigated[8].Chemical derivatives of metformin,such as HL271,induce comparable neuromodulatory effects,without any metabolic action,an indication that the drug effects may be only partially related to glucose homeostasis as is suggested in most of the experimental studies discussed on the following paragraphs[9].
Ouet al[10]designed an Alzheimer’s disease (AD) model in an effort to elucidate the anti-neuroinflammatory properties of metformin.APPswe/PS1ΔE9 mice underwent treatment with the biguanide,resulting into overall neuroprotective effects,with attenuation of spatial memory impairment,neural cellular proliferation,decreased local inflammation (both inflammatory cells and cytokines) of the brain cortex and the hippocampal region,as well as,reduced amyloid-β plaque deposition.The study results were attributed to drug-induced altered regulation of AMPK,mTOR,ribosomal protein S6 kinase,p65 and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways[10].
Type 1 and 2 diabetes,induced in animal models through streptozotocin and highfat diet respectively,have been linked to aberrant hippocampal neuroarchitecture with accompanying inflammation.Long-term metformin administration was shown to have a positive effect on hippocampal neural proliferation and memory function,despite the achieved hypoglycemic effect,a pathway mediated through interaction with insulin receptor substrate-1viaadenosine monophosphate (AMP) -kinase phosphorylation cascade activation[11].
Following a similar pattern of beneficial neural effects,on a diabetic rodent model where both memory and spatial recognition where evaluated with passive avoidance tasks and Y maze spontaneous alternation tests,metformin administration appeared to reverse the diabetes-induced functional decline[12].Passive avoidance assesses the capacity of test subjects to avoid certain choices linked to painful stimuli,by use of their previous memory of similar situations,while the Y maze trial recruits several neural compartments and reviews the tendency of a subject for exploring new pathways,a process inherently linked to cognition.The treatment-mediated effects were attributed to numerous metabolic effects including achievement of normoglycemia,upregulation of vascular endothelial nitric oxide production,attenuation of oxidative damage and increased anti-apoptotic potential.
On a study including subjects with non-dementia vascular cognitive decline with impaired glucose homeostasis,the efficacy of donepezil when combined with either metformin or acarbose was evaluated as to the possible achievement of functional improvement.Carotid artery intima-media thickness (CA-IMT),cognitive capacity and IR where assessed at baseline and at 12 mo.The metformin-donepezil group showed superiority in the functional tests administered,a fact that can be attributed to the slower CA-IMT increase and decreased IR indexes when compared to the acarbose group,allowing for better neural tissue perfusion and metabolic signaling,respectively[13].
One of the several pathogenetic mechanisms explored in relation to DE,among other neurodegenerative processes,is autophagy dysfunction,leading to tissueaccumulation of non-functional peptides,in the form of aggravates.Chenet al[14]attempted to elucidate the effect of metformin administration on the regulation misfolded polypeptide clearance,by treating diabetic mice with an eight-week regimen of intraperitoneal metformin and/or chloroquine.Neural capacity was evaluated by the Morris water maze (MWM) test,while the presence of aggravates or abnormal tissue architecture were examined by histological preparations and immunochemistry.Biguanide treatment had a positive overall effect with enhancement of autophagy,reduction of hyperphosphorylated tau proteins and improved cognitive functionality,when compared to the control group[14].
Different treatment regimens comprising of metformin and ursolic acid combined or as monotherapy,as well as gliclazide were used by Mouryaet al[15]in rodents with metabolic and cognitive impairment due to chronic restraint stress (containment for 2 h/d for 30 d).A total of 60 subjects were subdivided into 10 groups according to treatment protocol,with several metabolic parameters relative to cardiovascular function and IRs were observed.Behavioral and neurological performances were assessed by the MWM test.While insulin sensitivity and cognition were improved in all treatment groups,the most marked anti-inflammatory and neuroprotective effects were produced by the combination of metformin with ursolic acid,suggesting the existence of a synergistic effect between the two[15].
Metformin-induced neuromodulation has been studied on non-diabetic subjects as well,as is the case in the study of Fatemiet al[16],including a population of ovariectomized mice as the treatment group.Post-surgical subjects presented with cognitive impairment,anxiety disorders and reduced brain-derived neurotrophic factor (BDNF).Treatment with metformin had beneficial effects on behavioral dysfunction and BDNF reduction on both of the treatment groups (Group A:7 mg/kg and Group B:15 mg/kg).Reinforcing the idea that the neuroprotective effects of the biguanide class are not solely the result of metabolic normalization of glucose homeostasis[16].
As opposed to the aforementioned studies,Wennberget al[17]found no correlation between metformin or other anti-diabetic treatments and cognitive capacity.T2DM subjects with lack of functional impairment at baseline (n= 508) were followed-up for a mean duration of 3.7 years.Mild cognitive impairment (MCI) diagnosis was defined as difference equal to or greater than 1 standard deviation than the age-specific mean score of the general population on each test administered.The study population was divided into 4 groups according to treatment type as following:insulin monotherapy,metformin monotherapy,other oral agents as monotherapy or diet and exercise without pharmacological intervention.A universal lack of positive effect on cognition was observed among all groups,with patients on metformin treatment having higher rates of MCI diagnosis at follow-up.The latter was attributed to vitamin B12 reduction secondary to long-standing metformin administration.While the results are validated by the size of the study population and the numerous validated cognitive tests performed,notable limitations such as partial correction of treatment group differences despite covariate consideration and propensity score utilization and lack of B12 measurement should be taken into consideration when evaluating the research conclusions[17].
Correspondingly,a study conducted on C57BL/6 mice of different age groups yielded neutral results concerning the effect of metformin on metabolic parameters while a negative,age-dependent,impact was observed on both spatial memory and visual acuity of the test subjects.Treatment regimen comprised of 2 mg metformin/mL of drinking water,which is analogous to a human dose of 1500-2000 mg/d(when converted in a body-weight dependent manner),for three months[18].While,the contradicting results could be partially attributed to the short study duration there is further research with similar conclusions,in which metformin attenuated memory dysfunction in female subjects and amplified it in males,on an experimental model of AD[19].
The relationship between the class of biguanides and functional neural capacity remains unclear due to several relevant research projects with controversial results.At the same time,the underlying pathophysiological mechanisms by which metformin exerts its effects on neural tissue have not been,as of yet,entirely elucidated.While there appears to be a positive predilection towards the exploration of metformin administration as a form of neuroprotection,mainly due to its potency in altering a multitude of signaling pathways in the cell cycle,further research is needed in order to clarify whether it is truly efficacious in the clinical setting on patients with diabetesinduced cognitive decline.
Alpha-glucosidase inhibitors
Some of the main representors of the class of alpha-glucosidase inhibitors (α-GIs) are acarbose,miglitol and voglibose.Yanet al[20]administered acarbose to SAMP8 mice for a period of 6 mo.The study population was divided into 3 groups,including the acarbose group (n= 9,9-mo old),young (n= 11,3-mo old) and old controls (n= 8,9-mo old).An age-dependent cognitive decline was observed when the control groups were compared,while the acarbose group showed attenuation of this decline,accompanied by higher levels of insulin,insulin receptors and acetylated histone H4 lysine 8 (H4K8ac).The altered functional phenotype of the acarbose group (less memory impairment,improved spatial recognition) was attributed to both the changes in the concentration of insulin and its receptor and the H4K8ac increase.Higher levels of the latter have been linked to ameliorated long-term memory formation[20].
Since the data concerning the neurological effect of α-GIs is scarce,with no relevant research including miglitol or voglibose,safe conclusions cannot be currently drawn for their possible actions on neural tissue.
Sulphonylureas
As far as the class of sulphonylureas (SUs) is concerned,there appears to be a lack of relevant clinical studies discussing their effects on the homeostatic regulation of the nervous system.Given their mode of action,through binding on adenosine triphosphate-sensitive potassium channels and the subsequent activation of voltagegated calcium channels,their possible use for inducing and regulating neuroexcitatory potentials is an interesting perspective.Currently available research discussing the role of SUs in the setting of cognitive decline is centered on the use of glimepiride and glibenclamide.
Isholaet al[21]administered glimepiride on a rodent model of paraquat-induced Parkinsonism with subsequent functional and molecular assessment of the treatmentinduced changes.Sulfonylurea treatment attenuated oxidative stress and activation of inflammatory cascades in the neural tissue,while,simultaneously,improving the paraquat-induced memory dysfunction and cognitive performance on the rotarod,open field and Y-maze trials[21].
Glibenclamide has been shown to exert long-term protective properties on the hippocampal cortex in the setting of traumatic brain injury (TBI)[22].Moreover,the aforementioned exerted a beneficial effect when used on an experimental AD model,viaregulating the activity of the hypothalamic-pituitary-adrenal axis and alleviating AD-related mood-disorders[23].
Thiazolidinediones
Thiazolidinediones (TZDs) are peroxisome proliferator-activated receptor (PPAR)agonists,also widely known as glitazones,have been established to interact with the cell cycle and inflammatory cascade.
Pioglitazone was administered as monotherapy and in combination with simvastatin,on a model of lipopolysaccharide (LPS)-induced cognitive dysfunction secondary to amyloid deposition and inflammation.While LPS exacerbated neural oxidative stress,amyloid Aβ deposition,glutamate tissue-levels and memory impairment,both simvastatin and pioglitazone mitigated the changes.The subjects performance on both the neurobehavioral tests chosen (Y-maze and novel object recognition) did not differ significantly between the combination therapy or the monotherapy group for each treatment alone,a fact possibly explained by both the substances exerting their anti-inflammatory properties on the same pathway of NF-κB signaling[24].In a different study,pioglitazone was administered on subjects with LPS-induced febrile seizures and subsequent memory deficits.On the treatment groups,proinflammatory markers,such as tumor necrosis factor alpha (TNF-α) and interleukine-1β (IL-1β),along with oxidative stress were reduced in the hippocampal neural tissue,with accompanying partial resolution of memory impairment and cognitive dysfunction[25].Moreover,a meta-analysis performed by Caoet al[26],on the efficacy and tolerance of antidiabetic treatment as adjunct therapy on AD indicated that pioglitazone (15 to 30 mg) was the most beneficial agent (when compared to placebo) in improving cognitive capacity.
Kushwahaet al[27]have indicated the existence of a rosiglitazone-induced antiapoptotic effect on cerebral cortical tissue of high-fat-diet diabetic mice,for which the underlying mechanisms have not been clearly established.PPAR-γ mediated epidermal growth factor signaling appears to be the most probable pathway by which both glial and neural cells are affected.In a similar fashion,on a model of spontaneously hypertensive rats with consequent brain damage,rosiglitazone exerted a neuroprotective effect by mediating oxidative stress and affecting the levels of apoptotic cellular pathway mediators,independent of blood pressure correction[28].
Although the anti-apoptotic effects of TZDs on neural tissue are both supported by their mode of action and have been recreated in the experimental setting,there is a current lack of clinical correlation with the molecular findings.In order to establish the possible treatment benefits of this class in DE or other neuropathologic states,there is a definite need for further studying the performance of TZD-treated subjects on functional tests assessing both cognitive capacity and memory impairment.
Incretins
The two antidiabetic drug classes acting on the metabolic pathway of incretin hormones are glucagon-like peptide-1 receptor agonists (GLP-1 RA) and dipeptidyl peptidase-4 inhibitors (DPP-4i).GLP-1 is a hormone with multiple effects in the gut,pancreas and neural tissues,affecting processes such as gastric motility,appetite,insulin and glucagon secretion,while DPP-4 is the enzyme that deactivates it.
DPP-4 inhibitors
Sitagliptin,vilagliptin,saxagliptin,linagliptin and alogliptin are the current DPP-4is being used for treatment of T2DM[29].
APP/PS1 mice having been treated with sitagliptin (20 mg/kg for an 8-wk period)underwent neurofunctional assessment with the MWM test.The treatment group presented with ameliorated functional potential attributed to upregulation of BDNF and activation of tyrosine receptor kinase B (TrkB) signaling[30].Through similar mechanisms of BDNF and tyrosine hydroxylase upregulation,sitagliptin administered on a model of Parkinson’s disease moderated memory deficits,in addition to cellular density increase of dendritic spines in the CA1 region of the hippocampus[31].Male Wistar rats with cisplatin-induced neurotoxicity further confirmed the neuroprotective effect of sitagliptin on both the molecular level and motor-cognitive performance,accredited to attenuation of drug-induced cerebellar damage[32].
As far as vildagliptin is concerned,upon administration in an Alzheimer’s experimental model,the substance exhibited anti-apoptotic action in the hippocampal tissue with accompanying attenuation of memory deficits,changes associated with reduced tau phosphorylation and increased expression of neurotrophic proteins.An important mediator pathway and possible treatment target,identified in the above study,was that of phosphorylated protein kinase B/p-glycogen synthase kinase 3β(Akt/GSK3β)[33].The exact same treatment signature was observed when vildagliptin was used on a model of streptozotocin-induced T2DM with diabetes-related cognitive decline[34].Fibroblast Growth Factor 21 (FGF21) has shown superiority when compared to vildagliptin with the study therapeutic end-points being improvement of metabolic function and neuroprotection.Despite both the substances having insulinsensitizing,anti-apoptotic,mitochondrial and cognition-sparing properties,they differed on several other measurements.FGF21 was a more potent regulator of metabolic parameters and synaptic plasticity in the hippocampus[35].
Saxagliptin (0.25/0.5/1 mg/kg for 60 ds) has shown neuroprotective properties on streptozoticin-induced AD rats by increase of hippocampal GLP-1 levels,decrease of amyloid plaque formation and deposition[36].A slightly different rat model of AD disease,with cognitive deficits produced by D-galactose treatment,was used as grounds for comparing the efficacy of saxagliptin and metformin on learning and memory impairment secondary to aberrant insulin signalling.Several parameters on the MWM test were improved by antidiabetic treatment,along with oxidative biomarkers,tau phosphorylation products being normalized and insulin levels dropping with concurrent insulin receptor elevation[37].On the contrary,saxagliptin in an experimental model of Parkinson’s (produced by 6-hydroxydopamine administration) showed no cognitive- or motor-sparing properties but produced an interesting functional deterioration in the sham group,deeming it a possible candidate as post-traumatic stress disorder adjunct treatment[38].
Similar to other members of the DPP-4i class,linagliptin treatment has a beneficial role in ameliorating the progression of neural dysfunction on models of AD diseasevianumerous mechanisms such as amyloid plaque clearance,down-regulation of tau hyperphosphorylation,reduction of oxidative stress and mitochondrial dysfunction[39-41].In T2DM test subjects,the neuroprotective attributes of the substances have been linked to changes in cerebral perfusion.In one study,linagliptin treatment post-carotid inclusion related transient cerebral ischemia attenuated cerebral damage unrelated to glucose homeostatic regulation,by mediating oxidative stress and blood brain barrier permeability[42].Further,Hardiganet al[43]studied the effects of a 4-wk treatment regimen with linagliptin on vascular remodeling and flow properties of the middle cerebral arteries with beneficial effects being observed on the treatment group.The neuromodulatory role of linagliptin when compared to glimepiride is being studied by use of a composite 3-trial score (Mini-Mental State Examination,Trail Making Test,Verbal Fluency Test) in the cognition sub-study of double-blind,randomized Cardiovascular Safety of Linagliptin (CAROLINA) trial,including 4335 participants with T2DM[44].
Much like linagliptin,alogliptin has been shown to exert an effect on the architectural and functional integrity of cerebral vasculature.In a mice model of middle cerebral artery occlusion,the treatment group mediated the results of tissue ischemia and restored the defects of the blood brain barrierviaaltering the expression patterns of metalloproteinases and their inhibitors along with occludin and zona occludens-1 proteins[45].On another model of diabetic nephropathy with silent cerebral infracts,the combination of alogliptin and hyperbaric oxygen treatment had a beneficial restorative effect on neural function[46].Additionally,on high-fat fed doubly-negative apolipoprotein E mice with resultant cognitive decline,alogliptin upregulated BDNF and calcineurin hippocampal production with accompanying higher performance on MWM and novel object recognition test than controls[47].
GLP-1 receptor agonists
The currently approved GLP-1 agonists are exenatide,liraglutide,lixisenatide,albiglutide,dulaglutide and semaglutide.
In three studies (two included subjects with AD and one with T2DM) where the long-lasting GLP-1 analogue exenatide was used,restored BDNF signaling resulted into improved neurocognitive capacity by inhibiting neural apoptosis,in a manner analogous to that discussed on previous segments[48-50].Baderet al[51]used a sustainedrelease preparation of the substance,named PT302,in order to study the role of exenatide treatment in TBI.Subjects in the treatment group presented with a downregulation of pro-inflammatory markers in the neural tissue,prolonged cellular survival and reversal of functional impairment[51].Similarly,on the topic of TBI,Rachmanyet al[52]administered exenatide on a similar study population of mice with mild TBI,measuring both the neurofunctional changes and levels of synaptophysin (a biomarker for the viability of presynaptic neurons),pre- and post-trauma,with treatment ultimately attenuating the effects of the injury.Other changes following exenatide treatment include remodeling of hippocampal tissue architecture and diabetes-related deficits reversal,reduction of cortical TNF-α levels,preservation of brain choline acetyltransferase activity and improved amyloid oligomer clearance with subsequent decreased deposition[53,54].A novel dual incretin agonist with combined gastric intestinal peptide and GLP-1 activity,the latter in the form of exenatide,has shown similar neuroprotective actions like memory refinement and hippocampal neurogenesis and synaptic remodeling along with a positive metabolic profile[55].
While liraglutide has been shown to effectively attenuate memory and functional deficits in subjects with various AD or similar pathology patterns in neural tissue,through mediating tau hyperphosphorylation and amyloid deposition[56-58],contradicting research does exist,in which 12-wk liraglutide treatment was not superior in cognitive function improvement when compared to placebo[59].In the setting of cognitive decline following mood disorders,the GLP-1 RA improved performance in the Trail Making Test-B and composite Z-score of several neuropsychiatric scales measured,a change attributed to IR attenuation and other metabolic parameter modification[60].Post-treatment behavioral normalization was also noted in a study by Koshalet al[61]including mice manifesting with depression secondary to seizure activity.Some of the other changes in the treatment group were the reduction of oxidative stress and seizure activity[61].Cognitive-deficient rodents with T2DM treated with liraglutide presented with ameliorated functional potential as a result of activation and modification of downstream signaling pathways of AMPK,mTOR and phosphoinositide 3-kinase (PI3K)[62].The involvement of the mTOR pathway in the neuroprotective action of liraglutide was further confirmed in a study of streptozotocin-induced T2DM[63].
In a manner similar to other GLP-1 RAs,a pattern of reduced proinflammatory mediators and increased amyloid plaque clearance in APP/PS1/tau mice models of AD is observed with the administration of both lixisenatide and dulaglutide,resulting in improved neurocognitive potential.The pathways involved include those of p38-MAPK,protein kinase A and Akt/PI3K[64-66].Both the neuroprotective attributes of semaglutide and its superiority to liraglutide in improving cognition have been observed in mice models of Parkinson’s disease caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine[67,68].
SGLT-2 inhibitors
Sodium-glucose cotransporter 2 (SGLT2) inhibitors exert their actions on several tissue types,with their potency as antidiabetic substances stemming from their ability to hinder renal glucose reabsorption in the proximal tubule of the nephron.Members of this class currently in use are canagliflozin,dapagliflozin,empagliflozin,ertugliflozin,ipragliflozin,luseogliflozin,tofogliflozin with sotagliflozin,a dual SGLT1/SGLT2 inhibitor,in phase III clinical trials.The relationship between neural functional capacity and memory integrity and SGLT2 inhibition has been explored in studies utilizing canagliflozin,dapagliflozin and empaglilozin.
Arafaet al[69]studied the effects of canagliflozin treatment on memory dysfunction secondary to scopolamine administration.As an end-result of SGLT2 inhibitor treatment,neural tissue monoamine and acetylcholine levels were increased with M1 receptor activity,a biochemical shift culminating into improved cognitive function on MWM and Y maze trials[69].Similar patterns of altered acetylcholine signaling postcanagliflozin treatment were described on a similar study with diabetic rodents that included a metformin treatment group as well[70].
Dapagliflozin both as monotherapy and in combination with liraglutide has shown beneficial effects on memory and cognition,following remodeling of neural tissue with increased expression of doublecortin and synaptophysin (biomarkers of neural proliferation and synaptic formation respectively),as well as reduced IR[71].
The effect of empagliflozin on cognitive function was documented in the study by Linet al[72],after a 9-wk regimen on db/db mice.Assessment with the MWM test and immunohistological examination of cortical tissue was subsequently performed.The cognitive function of the treatment group was superior to that of age-matched controls,with concurrent attenuation of oxidative stress and increased BDNF levels[72].
Given the relative lack of data for this antidiabetic class,combined with the fact that the possible mediating mechanisms,either direct molecular or indirectviamodification of hemodynamic parameters,for their action on neural tissue have not been elucidated as of yet,there is definite need for further research on the subject.
Insulin
Neural tissue IR is an important substrate for the cognitive decline observed on diabetic subjects,especially in the hippocampal region.Numerous architectural and molecular changes fuel the pathologic process,including increased amounts of oxidative stress,activation of inflammatory cascades,peptide formation and aberrant deposition,commonly in the form of amyloid,as well as dysregulation of the hypothalamic-pituitary-adrenal axis[73].As would be expected,since the basis of diminished functional capacity in T2DM is formed on the existence of IR,treatment with insulin,in many forms,has proven to be beneficial in ameliorating the relevant pathophysiological alterations.
Several studies have emerged,exploring the use of insulinviaintranasal delivery,so as to bypass the blood brain barrier.This route allows for rapid achievement of therapeutic concentrations in the target tissue and treatment effectiveness,with accompanying cognitive improvement post-therapy[74-76].Some of the proposed mechanisms for explaining the attenuation of neurofunctional deterioration caused by T2DM include altered activation of electrolyte channels (mostly calcium-related),neuropeptide expression pattern differentiation,increased clearance of peptides(hyperphosphorylated tau and Aβ) that deposit as neurofilaments,synaptic remodeling and activation,upregulation of N-methyl-D-aspartate receptors turnover and improvement of hemodynamic parameters such as neural tissue perfusion[75].
On a study performed by Maimaitiet al[75],short-acting insulin lispro (Humalog)and long-acting insulin detemir (Levemir) were administered intranasally on a rat model of age-related mental impairment.Both the long- and short-acting compounds were equally effective in improving memory recall,matching the performance of aged members in the treatment group,to that of young rodents in the control group[75].Slightly different results came from the study of Benedictet al[77]where despite both regular and fast-acting insulin improving cognition when compared to the control group,the short-acting insulin aspart was more efficient than regular insulin in memory recall testing.
In a different research project,long-acting insulin analogs (glargine,detemir,degludec) were compared to regular insulin by use on cultured cortical neurons of rodents.Glargine,detemir and regular upregulated cortical BDNF,and activation of the Akt signaling cascade,with degludec having marginally inferior efficacy.Furthermore,regular and glargine ameliorated memory and cognition (as estimated by performance on the Y maze),showing superiority over detemir[78].
Finally,many of the physiological actions of insulin in the neural system are mediated by insulin-like growth factor-1 (IGF-1) receptors.Due to the aforementioned,similarly to insulin,use of neurostimulating factors with analogous activity on target tissues,such as IGF-1 has yielded promising results in the setting of neural proliferation and damage recovery post-trauma[79,80],neurodevelopmental disorders[81],neurovascular dysfunction[82]and IR[83].
Research data pertaining the use of insulin in the setting of cognitive decline,confirm the relationship between IR and mental deterioration,a state reversible by treatment with insulin or insulin-sensitizers.Further research could provide insight on the appropriate insulin delivery methods for achieving maximum therapeutic concentrations and treatment efficacy while minimizing risk,so as to fully utilize the potential of this therapeutic approach for diabetic and non-DE.A brief table containing all the aforementioned cognitive capacity experimental tests used on rodents is provided below (Table1).
CONCLUSION
DE is term describing a multifactorial state of neural dysfunction resulting from T2DM and its hallmark,IR.Current antidiabetic regimens appear to have a beneficial effect on cognitive decline and memory impairment secondary to diabetes and other causes.Most of the research data on the subject derives from studies on metformin,TZDs and incretins,with further elucidation being required for the role and mechanisms of sodium-glucose cotransporter inhibition on neural functionality.As has been shown by the intranasal delivery of insulin,the development of vectors allowing for direct access to the CNS without inhibition from the blood-brain-barrier could open up some very interesting perspectives for repurposing the antidiabetic therapy as means to effectively treat mental dysregulation states.Moreover,the extensive elucidation of the underlying pathophysiology allowing for oral antidiabetic medication to affect neural functionality could provide insight on the reasons behind cognitive impairment in T2DM,while also allowing for formulation of proper guidelines for hinderance of its development and ultimately,treatment.
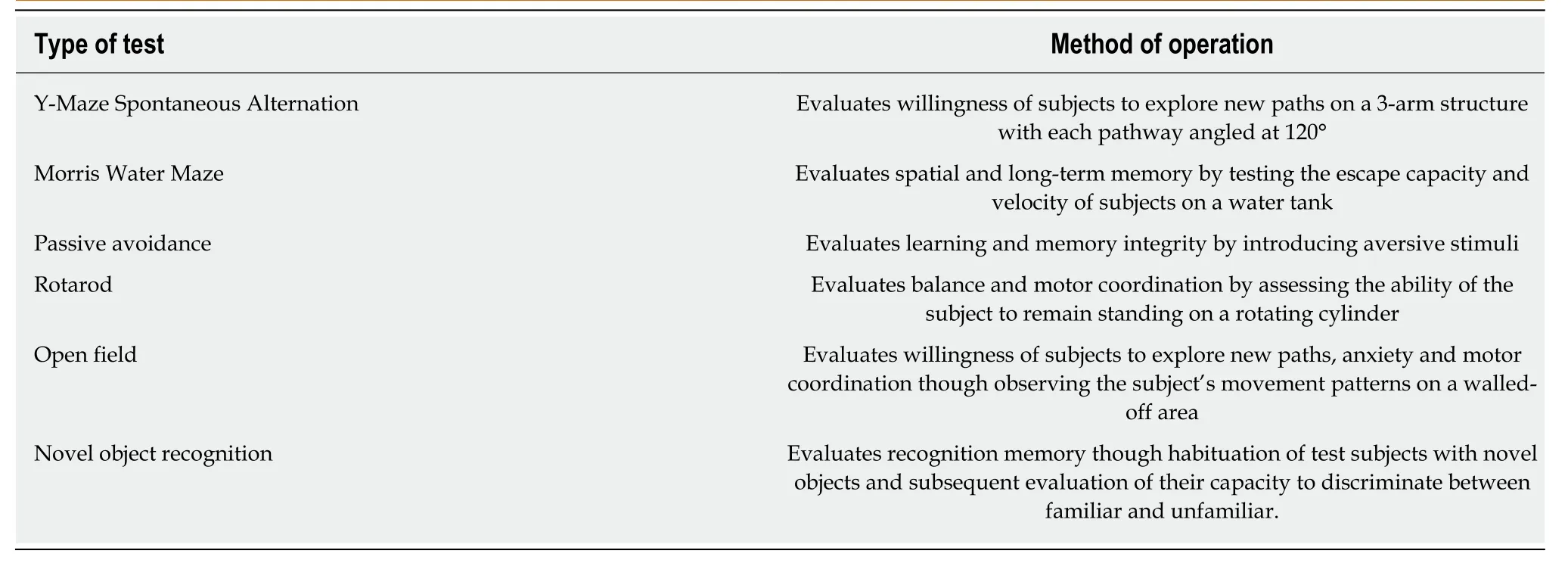
Table1 Experimental trials for the evaluation of cognitive capacity and memory impairment on rodent study populations
 World Journal of Diabetes2019年3期
World Journal of Diabetes2019年3期
- World Journal of Diabetes的其它文章
- Do we need to screen every patient in intensive care unit for diabetes in community with high prevalence of diabetes?
- Optimized health care for subjects with type 1 diabetes in a resource constraint society:A three-year followup study from Pakistan
- Burden of diabetic foot ulcer in Nigeria:Current evidence from the multicenter evaluation of diabetic foot ulcer in Nigeria
- Targeted genotyping for the prediction of celiac disease autoimmunity development in patients with type 1 diabetes and their family members
- Screening the RFX6-DNA binding domain for potential genetic variants in patients with type 2 diabetes
- Crosstalk between gut microbiota and antidiabetic drug action