
GC测定氢化可的松中甲醇、氯仿、1,2-二氯乙烷的残留量
2019-04-12 05:32:08白秀萍董玉杰
食品与药品 2019年2期
白秀萍,董玉杰
(1.淄博市食品药品检验研究院,山东 淄博 255086;2.山东金城生物药业有限公司,山东 淄博 255000)
氢化可的松为肾上腺皮质激素类的典型代表药物,可调节糖、脂肪和蛋白质的生物合成及代谢,具有抗炎、抗毒、抗休克及抗过敏等药理作用,既可作为糖皮质激素使用,也可作为制备其它甾体激素药物的起始化合物[1]。目前国内氢化可的松的制备均采用半合成方法,工艺中使用甲醇、氯仿、1,2-二氯乙烷等有机溶剂,限值见表1。为保证产品安全,本文依据中国药典2015年版四部及相关文献[2-3]建立了测定残留溶剂甲醇、氯仿、1,2-二氯乙烷残留量的气相色谱法。

表1 氢化可的松残留溶剂限度表
1 仪器与试药
6890N 气相色谱仪(配FID检测器,Agilent G1888 顶空进样器,20 ml顶空瓶,美国Agilent);氢化可的松(批号:0905034,0905035,0905045,0905046,山东新华);甲醇(分析纯,天津四友),氯仿(分析纯,国药集团),1,2-二氯乙烷(分析纯,莱阳经济技术开发区精细化工厂),二甲基亚砜(分析纯,天津福晨)。
2 方法与结果
2.1 供试液制备
2.1.1 空白溶液制备 精密量取二甲基亚砜1.0 ml置入20 ml顶空瓶,密封,待用。
2.1.2 标准贮备溶液制备 标准贮备溶液A:精密称取无水甲醇0.2 g(约0.25 ml)、氯仿10 mg(约7 μl)、1,2-二氯乙烷10 mg(约7 μl)置入已加入少量二甲基亚砜的100 ml量瓶溶解,用二甲基亚砜稀释至刻度,摇匀得A液。
标准贮备溶液B:取5.0 ml标准贮备溶液A,用二甲基亚砜稀释至100 ml,混合均匀。
2.1.3 标准溶液制备 精密量取标准贮备溶液B 1.0 ml置入20 ml顶空瓶,密封,混合均匀,同法配制6份。
2.1.4 样品溶液的制备 精密称取样品约0.1 g置入20 ml顶空瓶,加入1.0 ml二甲基亚砜溶解,密封。每批样品制备2份。
2.1.5 样品空白溶液制备 取样品(批号:0905034),研细,充分干燥,将其中残留溶剂降至较低的水平,作为样品空白。称取样品空白约0.10 g,精密称定,置入20 ml顶空瓶,精密加入二甲基亚砜1.0 ml溶解,密封,混合均匀。
2.2 仪器条件
毛细管色谱柱DB-624(30 m×0.53 mm,3.0 μm);柱温:初始温度40 ℃,保温10 min,以60 ℃/min升至200 ℃保持3 min;进样口温度:140 ℃;检测器温度:250 ℃;载气:N2;载气流速:5 ml/min;分流进样,分流比(1:1);平衡温度:120 ℃;平衡时间:30 min;针温:130 ℃;Transfer line 温度:140 ℃;加压时间:0.5 min;进样体积:1.0 ml。
2.3 计算方法
将空白溶液、标准溶液和样品溶液,依次置入顶空进样器,120 ℃下平衡30 min。取空白溶液进样,记录图谱,在标准溶液和供试溶液中扣除溶剂峰。取标准溶液进样。溶剂出峰顺序依次为甲醇、氯仿、1,2-二氯乙烷。相邻组分之间的分离度R,应不小于1.5;取6份标准溶液,连续进样,以3种溶剂峰面积计算的RSD,均应不大于15.0%。取每个供试溶液进样,各进样1次,记录图谱。计算公式如下。

注:X:残留甲醇、 氯仿、1,2-二氯乙烷的量(ppm);Ai:供试溶液的图谱中溶剂(i)的峰面积;A0:空白溶液的图谱中相应溶剂(i)的峰面积;Asi:标准溶液的图谱中溶剂(i)的峰面积;W:样品的称量(g);Wsi:每个溶剂的称重(g)。
2.4 专属性试验
取空白溶液进样,分析溶剂(二甲基亚砜)对检测的影响;取样品空白溶液进样,分析样品对检测的影响;分别取甲醇、氯仿、1,2-二氯乙烷各1 μl置入已加入1.0 ml二甲基亚砜的顶空瓶,密封,混合均匀,再分别进样,记录各溶剂的保留时间,用于各个溶剂峰的定性;取标准溶液进样,并记录各溶剂出峰顺序、保留时间,计算相邻溶剂峰的分离度R,判断各溶剂之间无干扰,则各相邻溶剂之间分离度R均应大于1.5;取样品空白0.10 g加入标准溶液1.0 ml溶解,再进样,若判断样品空白对残留溶剂分离度无影响,则各相邻溶剂峰的分离度R均应大于1.5,且与未加入样品时所得分离度无明显差异。
结论:标准溶液中出峰顺序依次为甲醇、氯仿、1,2-二氯乙烷;保留时间依次为1.939,6.442,8.017 min;空白溶液在各溶剂出峰时间均无干扰峰,溶剂(二甲基亚砜)对检测无影响;样品空白在各溶剂出峰时间均干扰峰,样品对检测无影响;分离度R甲醇-氯仿=24.3,R氯仿-1,2-二氯乙烷=6.2,均大于1.5,加入样品对分离度无明显影响,专属性符合要求。
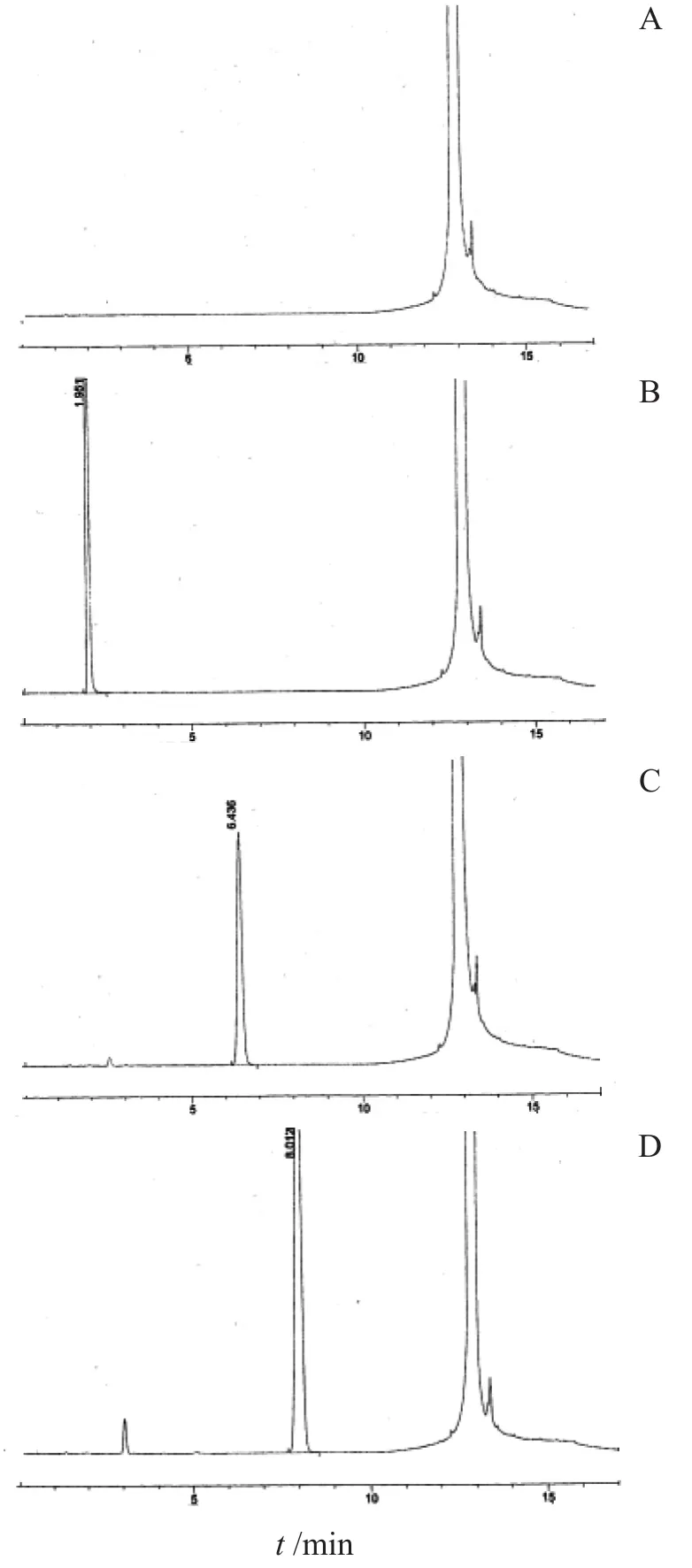
图1 专属性试验色谱图
2.5 系统适用性试验
称取甲醇0.1932 g、氯仿0.0105 g、1,2-二氯乙烷0.0100 g,按照方法描述配制标准溶液6份,连续进样。计算待测溶剂的理论塔板数N;连续进样6次,计算峰面积RSD。标准溶液中甲醇、氯仿、1,2-二氯乙烷的理论塔板数依次为3406,9063,11583;峰面积RSD依次为1.3%,2.3%和1.9%,均小于15.0%。方法系统适用性试验符合验证标准。
2.6 精密度试验
按照2.5 项下系统适用性试验数据,以每个溶剂峰面积的RSD评价方法的重复性,标准溶液重复进样6次,甲醇、氯仿和1,2-二氯乙烷峰面积RSD(n=6)分别为1.3%,2.3%和1.9%,均小于15.0%,方法的精密度良好。
2.7 线性关系试验
1,2-二氯乙烷标准贮备液:精密称取1,2-二氯乙烷0.0201 g置入已加入一定量二甲基亚砜的10 ml量瓶,用二甲基亚砜稀释至刻度,混合均匀。
标准贮备溶液A:精密称取甲醇0.3960 g、氯仿0.0246 g置入已加入一定量二甲基亚砜的100 ml量瓶,再加入1,2-二氯乙烷标准贮备液1.0 ml,用二甲基亚砜稀释至刻度,混合均匀。取标准贮备溶液A 0.25,0.5,1.0,1.5,2.0,2.5,3.0 ml分别置入100 ml量瓶,用二甲基亚砜稀释至刻度,混合均匀,得到系列浓度标准贮备溶液。
系列浓度标准溶液:从低浓度到高浓度,依次取系列浓度标准贮备溶液各 1.0 ml置入20 ml顶空瓶,密封,混合均匀,得到系列浓度标准溶液。每个溶液制备3份。
分别取上述系列浓度标准溶液,120 ℃平衡30 min,分别进样。以标准溶液中甲醇、氯仿和1,2-二氯乙烷的峰面积对其质量(以µg计,等于浓度×1.0 ml),使用Origin 5.0软件进行线性回归,得到每个溶剂标准曲线,线性方程及相关系数R。结果表明:甲醇在99~1188 ppm浓度范围内,线性方程为Y=7.718+8.861X,R=0.9999;氯仿在12.3~73.8 ppm浓度范围内,线性方程为Y=3.094X-0.153,R=0.9987;1,2-二氯乙烷在1.005~6.03 ppm浓度范围内,线性方程为Y=8.472X-0.080,R=0.9963。表明峰面积与浓度均呈良好线性关系。
2.8 准确度和回收率试验
样品空白的制备:取样品(批号:0905034),研细,充分干燥,将其中残留溶剂降至较低的水平。称取样品空白0.10 g,精密称定,置入20 ml顶空瓶,精密加入二甲基亚砜 1.0 ml溶解,密封,混合均匀得样品空白溶液。取2.7项下的No.2、No.6、No.7标准贮备溶液。标准溶液 取系统适应性试验项下的数据进行计算。
取20 ml顶空瓶9个,分为3组,每组3个,各精密称取样品空白0.10 g。分别取线性项下No.2,No.6,No.7标准贮备溶液各1.0 ml,分别加入到每组顶空瓶中,使溶解,密封,混合均匀,得到相当于限度20%,100%,120%测试溶液,测定。
计算方法 计算每个溶剂每个浓度的回收率,方法的回收率以3个浓度测定的回收率的平均值评价。计算公式如下。
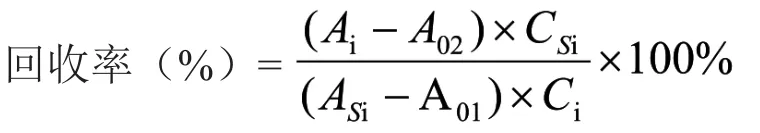
注:Ai:测试溶液图谱中溶剂(i)的峰面积;A01:溶剂空白图谱中溶剂(i)的峰面积;A02:样品空白图谱中溶剂(i)的峰面积;ASi:标准溶液图谱中溶剂(i)的平均峰面积;Ci:测试溶液中溶剂(i)的加入量,µg;CSi:标准溶液中溶剂(i)的量,µg。

表2 回收率试验结果(n=3)
以每个溶剂的回收率评价方法的准确度。结果见表2。甲醇、氯仿和1,2-二氯乙烷的回收率分别为98.5%,95.0%和98.1%,回收率的RSD分别为2.3%,2.5%和2.6%;每个溶剂的回收率均介于90.0%~110.0%,方法准确度良好。
2.9 检测限和定量限
取线性测试项下标准溶液逐步用溶剂稀释,得到适当浓度的溶液。在色谱系统下,分别进样,当待测组分的信噪比为2~3倍时,对应的浓度为该组分的最小检测浓度;当待测组分的信噪比为10~20倍时,对应的浓度为该组分的最小定量浓度。甲醇、氯仿和1,2-二氯乙烷的检测限分别为0.5 ppm,0.5 ppm和0.2 ppm,定量限分别为2 ppm,2.5 ppm和0.9 ppm。
2.10 耐用性
分别由两名化验员(化验员1和化验员2),使用两支同型号,不同色谱柱,改变色谱条件,柱温提高和降低5%,流速提高和降低5%,分别进行系统适用性验证。结果见表3。
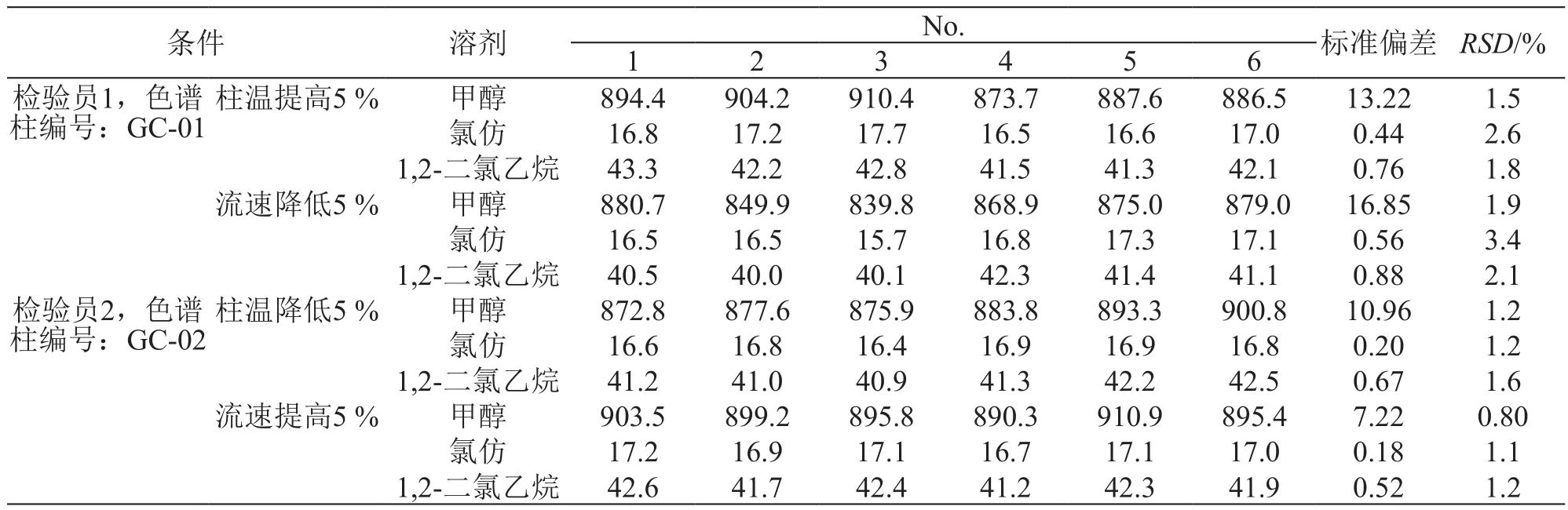
表3 耐用性实验结果
计算各溶剂峰面积的RSD(n=6),以RSD评价方法的耐用性。结果表明,不同化验员用使用不同色谱柱;提高和降低柱温,提高和降低流速,连续进样6针,RSD均小于15.0%,表明方法的耐用性良好。
3 样品检测
取4批氢化可的松样品(批号:0905034,0905035,0905045,0905046),按照规定条件检测。结果见表4。
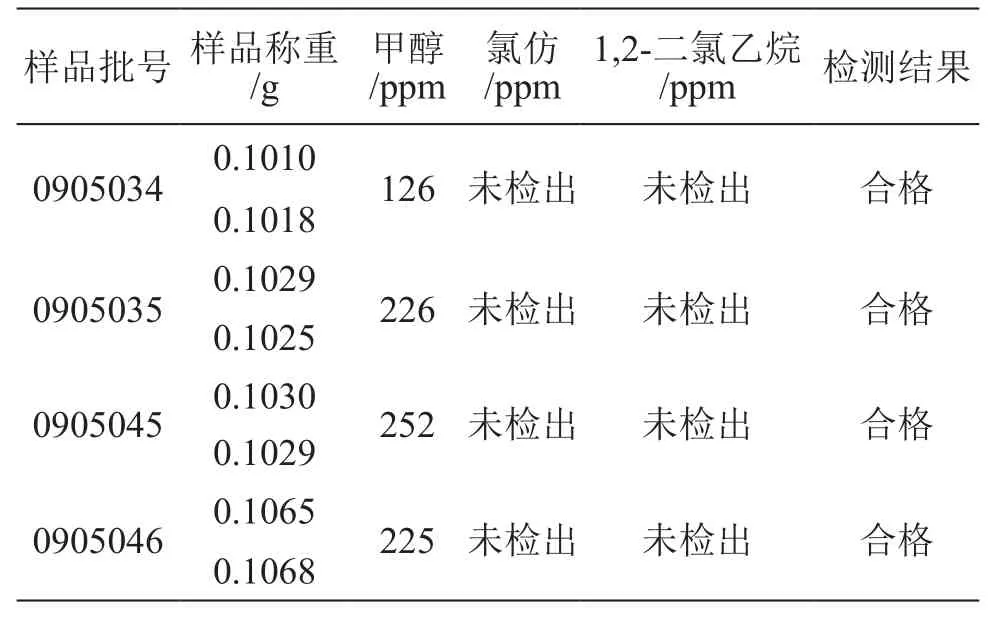
表4 样品检测结果
4 结论
本文参考中国药典2015年版四部及相关文献,采用气相色谱顶空进样法测定氢化可的松中甲醇、氯仿、1,2-二氯乙烷溶剂残留,考察了仪器条件、方法学研究,线性、准确度、精密度等均满足要求,建立了测定溶剂残留的检测方法,对4批样品进行测定。表明该法灵敏度高、精密度好,可用于氢化可的松中甲醇、氯仿、1,2-二氯乙烷溶剂残留的测定。
猜你喜欢
中国石油石化(2023年20期)2023-11-30 02:21:11
石油化工技术与经济(2023年5期)2023-03-11 22:39:09
阅读(中年级)(2022年9期)2022-10-08 01:56:14
小学阅读指南·低年级版(2021年4期)2021-04-20 03:56:27
化学与生物工程(2021年2期)2021-02-25 12:28:02
化工科技(2020年1期)2020-03-16 08:30:00
数学大王·中高年级(2019年5期)2019-06-09 10:20:52
安徽化工(2018年6期)2019-01-11 13:04:52
新高考·高一数学(2018年8期)2018-12-03 02:00:16
中国科技博览(2017年41期)2017-11-25 04:31:10
