
离子交换法改性NaY分子筛微球催化剂制备研究
2019-04-03 07:51豆高雅
陶瓷 2019年3期
豆高雅
(榆林市瀚霆化工技术开发有限公司 陕西 榆林 718100)
Y型沸石是用作催化剂和吸附分离剂的主要品种之一,应用于石油催化裂化工业过程,是20世纪60年代开始发展起来的一项新兴炼油技术。由于Y型沸石(特别是高硅Y型沸石)催化剂具有活性高、稳定性好等优点,己使整个催化裂化工业面貌大为改观[1]。Y型分子筛晶胞中有3种笼子:双六圆环笼(也称六方棱柱笼)、方钠石笼(也称β笼)和超笼(也称α笼)[2]。Y型沸石晶胞由18个四元环、4个六元环和4个十二元环所构成,有48个顶角[3]。其主空穴(超笼)的最大直径为1.25 nm,体积0.850 nm3,主孔道入口孔穴十二元环的直径为0.8~0.9 nm。Y型沸石作为FCC催化剂的活性组分,裂化反应主要在主孔道中进行。Y型分子筛晶体是在碱性体系合成的,初始晶体为Na型(NaY),NaY没有催化裂化活性,必须将绝大部分Na+用其它阳离子(H+、RE3+等)交换出来,变成HY、REHY、REY等才能呈现出固体酸特性,以作为裂化催化剂的活性组分[4]。
在NaY晶胞中,Na+分布在晶格的不同位置:S1(或称SI)、S2(或称SI')、S3(SII)、S4(SII')、S5(SⅤ)、U、SⅢ[5]。S1位Na+在体积最小的立方柱笼内,很难被其它阳离子交换出来,其它位置的Na+都能不同程度地被H+、NH4+、Re3+等交换出来,其中S3位的Na+移动性最强,最容易被交换出来[6]。
高岭土的基本组成是Al2O3·2SiO2·2H2O,典型NaY分子筛的基本组成是Na2O·Al2O3·4.9SiO2·9.4H2O。如果将天然高岭土进行活化处理,使其中的SiO2和Al2O3具有化学反应活性,然后加入适量NaOH进行水热晶化,可望以廉价的高岭土为原料合成出NaY分子筛,经进一步处理得到FCC催化剂。因为高岭土的主要物相组成是晶体高岭石,结构十分稳定,化学活性很低,需要将晶体破坏,以产生具有化学反应活性的活性SiO2和活性Al2O3,实现这一目标的简捷途径是将高岭土进行高温焙烧,再以焙烧高岭土为原料合成NaY分子筛。
1 实验部分
1.1 试剂与仪器
1.1.1 实验所用主要试剂(见表1)
表1 实验主要试剂
1.1.2 实验所用主要仪器(见表2)

表2 实验主要仪器
1.2 喷雾微球样品的制备
1.2.1 配制高碱偏铝酸钠
将一定量的蒸馏水移入三颈烧瓶中,冷凝回流。迅速称量氢氧化钠置于三颈烧瓶内,搅拌、加热并溶解。温度升至90 ℃后,加入定量的氢氧化铝,继续搅拌升温。当温度升至110 ℃,约5 min后,体系由混浊变为澄清,恒温搅拌2 h,移出溶液,密封备用。
1.2.2 导向剂的合成
利用自制水玻璃和偏铝酸钠溶液,按照实验所需配比配制导向剂。将水玻璃加入到三颈烧瓶中搅拌,缓慢加入偏铝酸钠,约15 min后,体系由有透明变为乳白色。继续搅拌30 min后,移至32 ℃恒温水浴16 h。老化结束后导向剂应立即使用,剩余的导向剂则在0~4 ℃冷藏保存,否则该导向剂失效。老化温度和时间对导向剂的质量都有重要影响,提高老化温度,则需相应缩短老化时间。
1.2.3 喷雾微球的制备
将原料按蒸馏水、水玻璃、高岭土、导向剂的顺序加入打浆釜,搅拌2 h后过筛,得细浆液。用TCIP2000中型喷雾干燥塔喷雾成形微球。根据喷雾浆液粘度及液固比不同,选择不同的喷雾塔操作条件:进风温度为300~330 ℃、进气口压力为0.15~0.3 MPa、进料速度为50~80 rpm。筛选出20~150 μm中间组分,作为制备催化剂的喷雾微球。
1.3 焙烧微球的制备
将喷雾微球在空气中高温焙烧,以破坏高岭石晶体结构,使部分转化成具有化学反应活性的无定形SiO2和Al2O3,另一部分转化为催化剂的基质,焙烧时间为2 h,料层厚度为2~4 cm。将喷雾微球分别在960 ℃(髙土)及850 ℃(偏土)焙烧,焙烧后的微球简称焙烧微球。
1.4 晶化微球的制备
晶化微球的制备流程如图1所示,在此制备工艺中,每一步骤都得到了不同特性的微球。
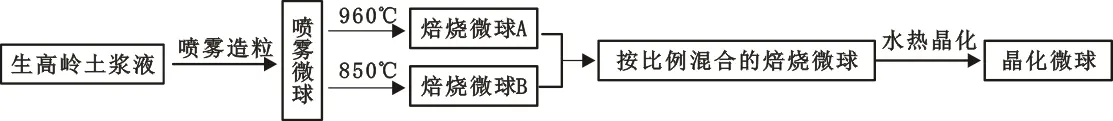
图1 晶化微球制备流程
将去离子水、NaY导向剂、硅酸钠等液体按计量投入平底烧瓶中,搅拌下投入焙烧微球,升温到92 ℃恒温晶化,视实验目的不同,晶化时间在12~32 h之间选择,本实验晶化时间选取24 h。晶化过程中,晶化体系的水分会蒸发损失,所以要实时均匀补充去离子水,以确保晶化过程的H2O/Na2O比不变。晶化结束后,用倾滤法除去母液,母液留存备用;滤饼用5~7倍(质量比)去离子水反复洗涤、过滤,直到滤液pH值达到10.5以下,收集滤饼备用。
晶化实验投料比见表3。

表3 晶化实验投料比
经上述处理得到的含NaY分子筛的微球称为晶化微球,是制备FCC催化剂的前躯体,也称前躯体微球。
1.5 离子交换与成品催化剂制备
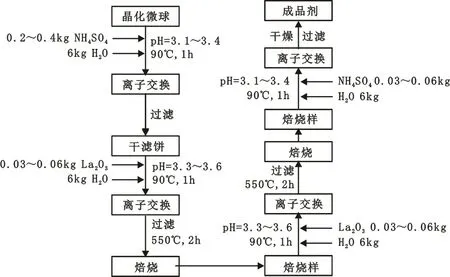
图2 成品剂制备流程
裂化催化剂是由晶化微球经4次离子交换和2次焙烧制得。其制备工艺流程如图2所示。
沸石分子筛FCC催化剂属于固体酸催化剂,它的酸性来源于氢离子(如HY)、交换态阳离子的分解(如NH4Y)、多价阳离子在脱水时的水解(如LaY)。由于Y型分子筛晶体是在碱性体系合成的,初始晶体为Na型(NaY),NaY没有催化裂化活性,必须将绝大部分Na+用其它阳离子(H+、RE3+等)交换出来。
晶化产物的后处理包括离子交换和焙烧两个工艺步骤。离子交换反应是可逆的,故必须进行多次交换才能达到较高的交换度。溶液的浓度、交换的次数、交换时间、交换温度等因素对钠的交换率都有影响。另外在离子交换过程中,位于小笼中的钠离子一般很难交换出来,进行中间焙烧,使残留的钠离子重新分布,移入易交换的位置,然后再用其它正离子交换,可以较大的提高交换度。
离子交换的目的是用NH4+和稀土离子RE+替代分子筛上的Na+,焙烧的目的是将水合RE+脱水,减小RE+在分子筛孔道中扩散阻力,通过高温固相扩散,进入分子筛孔道内部,与Na+发生固相交换和迁移。
第一次离子交换和第四次离子交换采用(NH4)2SO4为交换剂,加入量为干基CRM的20%~30%;第二次交换和第三次交换采用RECl3(以LaCl3)为交换剂,本实验用磷酸氢二铵进行补交。离子交换体系的液/固比值控制在5~6,体系pH为3.1~4.5,交换时间为60 min,温度为85~95 ℃,在交换过程中随着分子筛上Na+逐渐进入液相,体系的pH值会逐渐升高,应通过加入少量稀盐酸调节到所需要的pH范围,调节过量时用稀氨水溶液回调。每次离子交换结束后,除去交换母液,同时还用去离子水洗涤。
1.6 催化剂各成分含量分析
1.6.1 氧化钠含量测定
采用火焰光度计测定催化剂中氧化钠含量,其测定原理为:待测样品在被火焰雾化及燃烧过程中,其含有的Na+被激发后发射出的特征光谱线被仪器中滤光片分离后,投射于光电池上并被转化为光电流,而光电流的强度与待测样品的Na+浓度成正比,从而计算出氧化钠的含量。
1.6.2 稀土含量测定
在pH值5~5.5范围内,采用二甲酚橙做指示剂,六次甲基四胺作缓冲溶液,用EDTA标准溶液将稀土待测液由紫红色滴定至亮黄色,记下此时消耗的标准EDTA溶液的体积,从而计算出稀土待测液中稀土含量。
1.6.3 氧化铝含量测定
采用EDTA络合返滴定法测定催化剂中氧化铝的含量。其测定过程为:在常温下先用酒石酸掩蔽铁、钛及稀土等离子干扰,然后向弱酸性体系中加入定量的EDTA溶液,使其与铝离子生成稳定络合物,最后以二甲基橙为指示剂,在pH=5~6的条件下,用氯化锌标准溶液返滴定过量EDTA,计算出与氧化铝反应EDTA的量,即裂化催化剂中氧化铝的含量。
1.7 催化剂酸特性测定
采用程序升温脱附法(TPD)测定催化剂酸特性,以NH3为探针气体,用热导法测定其脱附速率和脱附量。实验以N2为载气,将一定量的样品置于U型石英管中,室温下通入NH3;待催化剂饱和吸附NH3后(约30 min),关闭NH3进气阀,用N2吹扫管路中剩余吸附气路至排气口无NH3逸出为止。吸附炉升温至100 ℃,等基线稳定后,开始持续升温,升温速率为10 ℃/min,用热导池测试NH3的脱附曲线。
1.8 活性测定
采用上海奥轮催化剂有限公司FCC催化剂微反活性(MAT)专用测定仪,原料为标准轻柴油。催化剂老化条件为800 ℃±1 ℃、100%水汽环境老化,老化时间采用国内通用的4 h、10 h和17 h。MAT反应器催化剂装量为5.000 0g±0.002 g,进油时间为70 s±1 s,进油量为1.56 00 g±0.002 g,反应温度为460 ℃±1 ℃,剂油比为3.2。反应产物用上海计算机所GC-112型模拟蒸馏专用色谱仪测定。
1.9 固定流化床评价

表4 固定流化床测定条件
采用北京拓川小型固定流化床裂化催化剂评价装置,分析评价了催化剂HD-C和工业对比剂QD的选择性,其过程为:在相同反应条件下(见表4),以减压渣油为原料,对经流化催化裂化反应后产生的汽油、柴油、干气、富气、焦炭等物质的生成量及组成,用气相色谱等仪器进行分析测试计算。
2 实验结果与讨论
2.1 结晶度与离子交换度分析
2.1.1 晶化产物结晶度分析
晶化体系反应硅钠比是指参与晶化反应的SiO2和Na2O的物质的量之比。在设计晶化方案时,晶化体系中总SiO2和总Na2O的量是定值,其由两部分组成,一部分是指参与晶化反应的SiO2和Na2O,主要涉及形成Y型分子筛骨架和沉积于微球表面的SiO2和Na2O,留存于固相中;另一部分是未参加反应和由于微球骨架被破坏而由固相中进入液相的SiO2和Na2O,留存于液相中(又称为晶化母液)。
由此可见,晶化母液主要由SiO2和Na2O组成,即稀水玻璃,其中SiO2来源于晶化体系中的水玻璃、高土及偏土;Na2O来自于水玻璃和NaOH溶液。所以,通过测定晶化母液中的SiO2和Na2O的含量,即可得知参与形成分子筛骨架的SiO2和Na2O的量,即可得出CRM的NaY含量(结晶度)与晶化体系中参与反应的硅钠比的关系。
利用上述原理,建立了结晶度(Y)与反应硅钠比(X)的标准曲线;通过标准曲线可以方便的查询出所制得的CRM的结晶度。此方法具有投资小,可快速查询分子筛结晶度等优点;其缺点是微球的中位径(d50)必须在55 μm左右且存在一定误差。
2.1.2 晶化体系反应硅钠比与结晶度的关系
为了考察晶化体系反应硅钠比与结晶度的关系,笔者计算出了晶化设计方案中总SiO2和总Na2O的量并且测出了晶化母液中SiO2和Na2O的量;然后将两者结果作差,得出参与反应的SiO2和Na2O的量,从而得出反应硅钠比(X)与结晶度(Y)关系表,见表5。
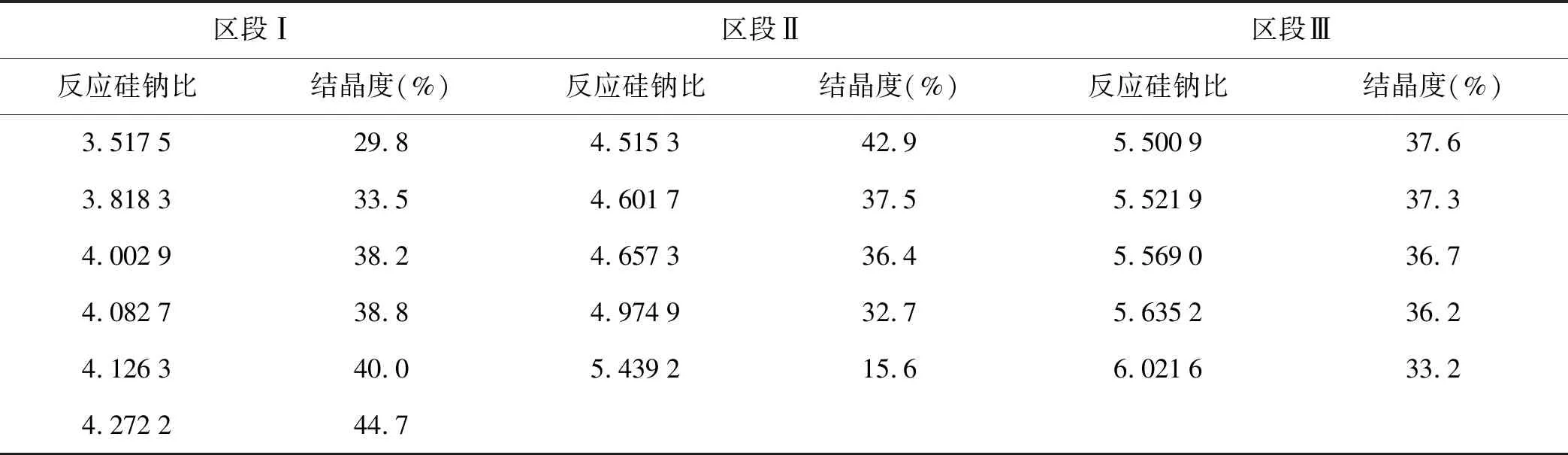
表5 反应硅钠比(X)与结晶度(Y)关系表
由表5可见,在反应硅钠比值3.517 5~4.272 2区域内,结晶度随反应硅钠比的升高而增大,当反应硅钠比值为4.272 2时,结晶度达到最大值44.7%;随着反应硅钠比继续升高,结晶度值在4.515 3~5.439 2区域内迅速降低,当反应硅钠比值超过5.439 2时,结晶度随之升高,但随着反应硅钠比的继续增大,结晶度值缓慢下降。
综上所述,CRM结晶度随反应硅钠比的升高,其呈先增大,后迅速减小,然后再增大,随后又缓慢减小的规律。因此,将晶化体系的反应硅钠比值按结晶度变化规律由小到大分为(Ⅰ)3.517 5~4.272 2、(Ⅱ)4.515 3~5.439 2和(Ⅲ)5.500 9-6.021 6三个区段并绘制成标准曲线。
2.1.3 区段I中反应硅钠比与结晶度关系
为了考察区段I中反应硅钠比与结晶度的关系,将区段I的反应硅钠比和结晶度的数据绘制成图,将图中数据点进行曲线拟合得到了其标准拟合曲线(见图3)。
由图3可见,反应硅钠比在3.5~4.2之间,随着其数值的增大,CRM结晶度也在增大,在反应硅钠比增大到4.2时,CRM结晶度已增大至40%,说明反应硅钠比对CRM的结晶度有较大影响。
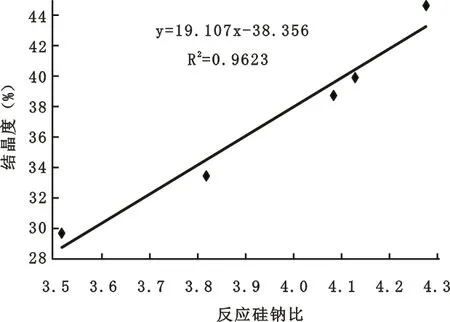
图3 区段I中的反应硅钠比和结晶度的关系
2.1.4 区段II中反应硅钠比与结晶度关系
为了考察区段II中反应硅钠比与结晶度的关系,将反应硅钠比和结晶度的数据绘制成图,并作出其标准拟合曲线(见图4)。
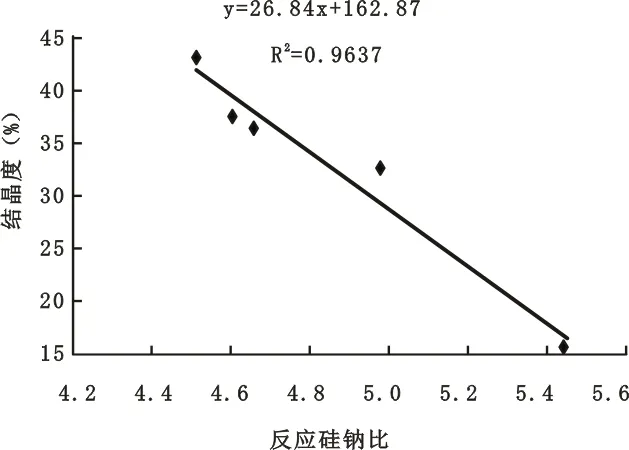
图4 区段II中的反应硅钠比和结晶度的关系
由图4可见,反应硅钠比在4.2~5.5之间,随着其数值的增大,CRM结晶度呈下降趋势,当反应硅钠比增大至5.5时,其结晶度可降至15%左右。
2.1.5 区段Ⅲ中反应硅钠比与结晶度关系
为了考察区段Ⅲ中反应硅钠比与结晶度的关系,将中区段Ⅲ的反应硅钠比和结晶度的数据绘制成图,并作出其标准拟合曲线(见图5)。
由图5可见,反应硅钠比在5.5~6.1之间,随着其数值的增大,CRM结晶度呈下降趋势,当反应硅钠比增大至6.1时,其结晶度已降至33%左右。
再由图5可见,区域II的拟合方程的斜率大于区域Ⅲ的,说明微球的结晶度在区域II中下降速率快,数值波动较为严重且受反应硅钠比影响最为敏感,说明晶化反应不稳定,因此,在设计晶化反应时,反应的硅钠比应适当避开此区域。
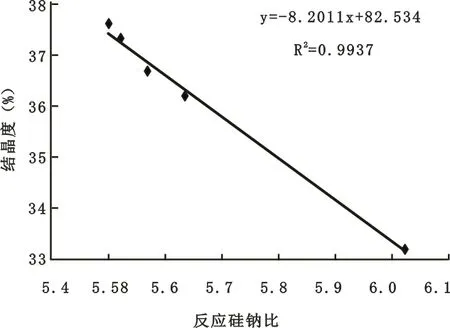
图5 区段Ⅲ中的反应硅钠比和结晶度的关系
晶化反应过程中并不是反应硅钠比越高越好,且随着反应硅钠比的增大,即SiO2含量的增高,CRM的结晶度并未一直增大,推测液相中的SiO2多以无定形态沉积于微球表面上,从而阻断了微球的晶化过程的进行。可得出如下结论:
1)在反应的硅钠比为3.5~4.2时,晶化微球的结晶度随反应硅钠比的增大而上升,其数学模型为Y=19.107X-38.356。
2)在反应的硅钠比为4.2~5.5时,晶化微球的结晶度随反应硅钠比的增大而下降,其数学模型为Y=-24.8765X+152.9557。晶化微球在此区域数值波动较为严重且受反应硅钠比影响最为敏感,说明晶化反应不稳定,因此,在设计晶化反应时,反应的硅钠比应适当避开此区域。
3)在反应的硅钠比为5.2~6.1时,晶化微球的结晶度随反应硅钠比的增大而下降,其下降速率较区域II中缓慢,其数学模型为Y=-8.2011X+82.534。
4)晶化反应过程中并不是反应硅钠比越高越好。随着反应硅钠比的增大,即SiO2含量的增高,CRM的结晶度并不会一直增大,此现象可能是由于液相中的SiO2多以无定形态沉积于微球表面上,阻断了微球的晶化过程而造成的。
本次晶化反应的反应硅钠比为3.8021,在区段Ⅰ内,代入数学模型求得结晶度,其结果见表6。

表6 NaY分子筛结晶度
2.1.6 交换产物分析
离子交换反应是可逆的,故必须进行多次交换才能达到较高的交换度。溶液的浓度、交换的次数、交换时间、交换温度等因素对钠的交换率都有影响。另外在离子交换过程中,位于小笼中的钠离子一般很难交换出来,进行中间焙烧,使残留的钠离子重新分布,移入易交换的位置,然后再用其它正离子交换,可以较大的提高交换度。将晶化产物用硫酸铵交换,交换后的微球经过滤、水洗、再过滤得一交料;将一交料用氯化镧(其成分见表7)溶液交换,经过滤、水洗、再过滤得二交料;将二交料在一定条件下进行焙烧,得一焙料;一焙料用氯化镧溶液重复交换一次即得三交料。将三交料在一定条件下进行焙烧得二焙料。二焙料用硫酸铵补充交换一次,交换产物经过滤、洗涤、过滤、干燥后得成品催化剂。为了提高钠的交换度,本实验用磷酸氢二铵进行补交。
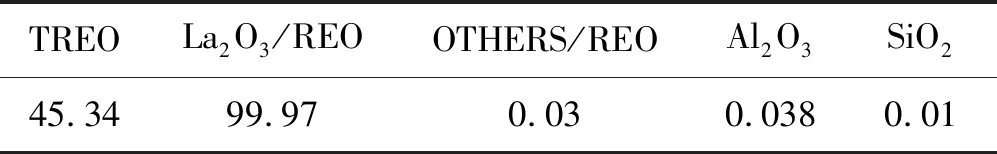
表7 氯化镧的化学成分(质量%)
2.1.6.1 钠含量分析
钠标准曲线如图6所示。

图6 钠标准曲线
由火焰光度计法换算母液中钠离子含量,进而求得催化剂中钠含量以及交换度(见表8)。
从表8中可以看出:

表8 离子交换过程中各阶段理化数据分析表
1) 微球的Na2O的含量和总交换度随着交换的过程的进行而明显降低。
2) 一交的Na2O交换度最高,这是因为一交前NaY中Na2O主要是存在分子筛超笼中,较易交换,故交换度最高。
3) 二交的Na2O交换度提高幅度减小,是因为二交前的物料Na2O主要存在于β笼中,不易交换出来,从而交换度较低。所以在三交之前必须先进行焙烧,使水合稀土离子进行脱水,以减小水合稀土离子半径,便于扩散到分子筛孔道内部;另一方面,使钠在焙烧的过程中进行固相迁移,在分子筛内部重新分布,迁移到比较容易交换的位置中,从而提高三交钠的交换度。同样,在四交之前再次进行焙烧,提高四交钠的交换度,使成品催化剂的Na2O含量达到工业催化剂的要求。
2.1.6.2 La2O3、Al2O3含量分析
RE2O3的含量随着离子交换过程的进行,含量逐步提高,RE2O3的含量达到4.13%,而在补交过程中稀土含量略有下降,说明在补交过程中稀土出现反交换现象。各交换过程稀土含量见表9。

表9 交换过程稀土含量
由表9可知,Al2O3的含量随着离子交换过程的进行,含量有所降低,是因为部分Y型分子筛在交换的过程中,破坏晶体结构,晶格上的铝被取代,进入液相,从而使Al2O3含量降低。离子交换是在pH小于5的酸性介质中进行的,这部分Al2O3会部分溶解进入交换液,从而微球的Al2O3会降低,说明晶化微球在交换和焙烧过程中Y型分子筛晶体遭到一定程度的破坏,导致随着离子交换的过程的进行,交换料的结晶度下降,说明部分Y型分子筛晶体在晶化微球后处理过程中有不同程度的崩塌,转变为无定形Al2O3和SiO2。
2.2 催化剂理化性质及性能评价
2.2.1 离子交换改性分子筛的性质
2.2.1.1 晶化微球的性质
将反应溶液按一定投料比例和投料顺序与焙烧土球混合搅拌,利用原位晶化工艺制备了NaY晶化微球。其性质为:结晶度为34.3 %,硅铝摩尔比为4.98,磨损指数为1.8 %,比表面积为420.3 m2/g,孔体积为0.36 mL/g。
2.2.1.2 磷改性后催化剂的物化性质
NaY沸石经铵和稀土离子改性,部分铵离子和稀土离子与Y型沸石超笼中钠离子发生交换作用,进而在水热处理过程中迁移进入方钠石笼中,再通过焙烧处理,使得稀土离子脱水进入β笼中,定位于β笼中的稀土离子能与沸石骨架的氧原子相互作用,形成六配位结构的配合物,提高了沸石骨架的稳定性,抑制了沸石在高温作用时的骨架脱铝,保护了沸石表面的结构酸羟基。
通过磷改性处理,使部分P进入沸石的骨架上,另一部分与稀土发生相互作用形成超细的RE-P-O复合氧化物,覆盖在沸石的外表面上,并占领部分酸性活性中心,同时部分磷还会与沸石表面的铝羟基作用形成酸性较弱的P-OH,这样Y型沸石的孔道内部经过充分的处理,酸性密度和强度得到优化,更多的原料烃分子吸引到孔道内部进行反应。
磷改性高岭土型催化剂的理化性质见表10。由表10可见,原位晶化微球的孔体积为0.34 mL/g,而经过磷改性后,孔体积增大到0.38 mL/g。微球在酸性环境中经过改性处理,有效地清除了孔内的堵塞物,导致孔体积增大。由表10还可看出,微球改性前后比表面积变化不大,表明改性和水热处理的条件较为缓和,对沸石的破坏程度很小。

表10 HD-C催化剂的理化性质
2.2.2 催化剂性能评价
2.2.2.1 相对结晶度测定
图7是本实验室制备的催化剂HD-C与LB-2催化剂的XRD分析图。
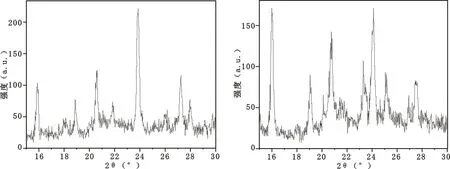
(a) HD-C的XRD图 (b) 对比剂QD的XRD图
图7 HD-C和对比剂QD的XRD图
表11是以Y型分子筛(533)衍射峰推算的催化剂上分子筛结晶度。

表11 催化剂的相结结晶度
注:a)为以(533)衍射峰推算的相对结晶度。
从表11看出,原位晶化催化剂的相对结晶度在34%,大于对比催化剂22%。
2.2.2.2 孔结构
表12是催化剂HD-C与对比剂QD的比表面、孔体积。从表12可以看出,催化剂HD-C的比表面积及孔体积大于对比剂QD,从比表面和孔体积角度说明催化剂HD-C对反应物分子有更好的吸附、扩散和可接近性。

表12 催化剂的比表面与孔体积
2.2.2.3 催化剂酸强度
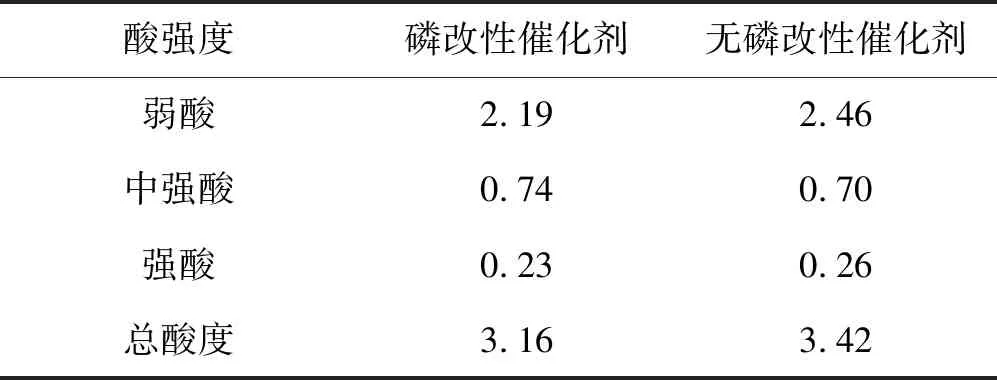
表13磷改性和无磷改性高岭土型催化剂的表面酸性的结果(mmol·g-1)
酸强度磷改性催化剂无磷改性催化剂弱酸2.192.46中强酸0.740.70强酸0.230.26总酸度3.163.42
采用NH3-TPD法测量催化剂的表面酸性,当碱性气体分子与固体酸性催化剂接触时,除了发生气固相物理吸附外,碱性分子还在催化剂酸性部位上产生化学吸附,这种化学吸附作用先从催化剂的强酸部位开始,逐步向弱酸部位扩展,脱附过程与此相反。由于氨分子既可以与催化剂表面B酸反应生成NH4+,又可以与L酸部位作用形成配合物,因此氨脱附法测得的酸量是包括B酸中心数目和L酸中心数目的总和。表13列出了磷改性和无磷改性催化剂表面酸性的结果。
表13结果显示,磷对催化剂的改性可以有效地调整催化剂的酸量和酸强度,磷的引入降低催化剂的总酸量、弱酸量和强酸量,可增加中强酸的酸量。
高岭土型催化剂的Y型沸石和基质几乎都是由高岭土经特殊工艺转化而成,由于催化剂强酸主要来自其中Y型沸石的B酸中心,而弱酸中心主要来自晶化反应的碱抽高岭土中的Al2O3的L酸中心和分子筛的弱酸中心。
当用含磷溶液处理样品时,磷酸盐水解生成磷酸,磷酸与样品的B酸中心反应时,OH-被H2PO4-取代,即磷羟基取代了硅铝羟基,由于两者的电负性相近(OH-为4.30,H2PO4-为4.38),桥羟基变成了端羟基,因而磷的引入导致其强酸中心数下降,酸强度降低。而磷酸与Al2O3的L酸中心发生的反应时,在磷含量较低时,样品表面羟基较多,易和磷酸形成多键及二键结构,由于多重键的形成,将会减少可利用的表面羟基及暴露的铝原子数目,使得样品Al2O3酸量及酸强度下降,综合磷对高岭土型催化剂中沸石和基质两方面的作用,催化剂的总酸量、弱酸量和强酸量降低,中强酸的酸量增加。

(a)催化剂HD-C (b)对比剂QD
图8催化剂的NH3-TPD图
图8是两种催化剂的程序升温氨脱附(NH3-TPD)图,主要用于鉴别催化剂的固体酸强度。
对比图8(a)和图8(b)的程序升温热脱附测试图可发现:
1)催化剂HD-C在175 ℃和200 ℃处的吸附特性与对比剂QD相近;
2)两种催化剂在300~400 ℃时有较大差别,HD-C的吸附量大于对比剂QD;
3)HD-C催化剂的总算量(TPD图积分面积)大于对比剂QD。
2.2.2.4 催化剂活性
为了考察催化剂的微型反应活性指数及水热稳定性,在微反评价装置(MAT)上做了相应的测试,表14是两种催化剂在800 ℃/100%水蒸汽不同老化时间的微活指数。
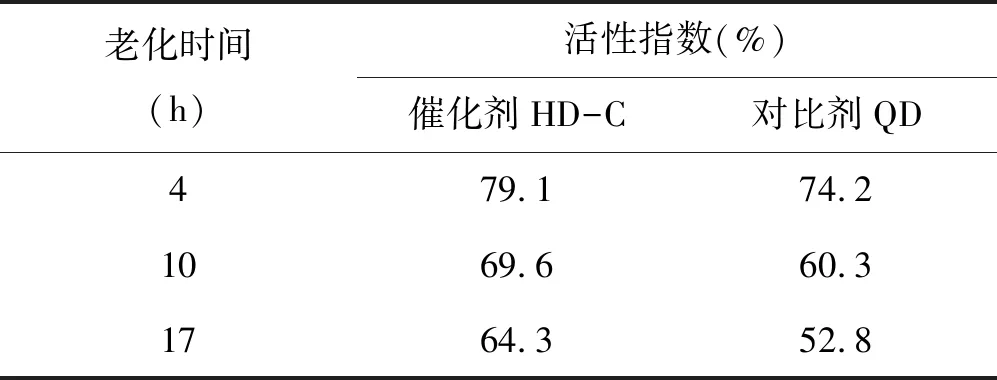
表14 催化剂的不同老化时间微活指数
注:800 ℃,100%水蒸汽
从表14中可以看出:
1)催化剂HD-C不同老化时间的微活指数均高于对比剂QD,具有较高的催化活性。
2)催化剂HD-C经17 h老化微活指数下降14.9%,而对比剂QD微活指数下降28.8%,说明催化剂HD-C具有良好的水热稳定性,随老化时间的增加微活指数的下降速度较慢,所以,催化剂HD-C的水热稳定性优于对比剂QD。
2.2.2.5 抗重金属污染能力
1)裂化催化剂镍中毒机理。沉积在FCC平衡催化剂表面的镍主要以NiAl2O4和Ni2O3的形式存在,也有少量NiO,Ni可部分替代H进入分子筛晶格中,从而降低分子筛酸性中心的酸强度,Ni亦可与非骨架铝作用形成Ni的铝酸盐,减小非骨架铝的数目,这将影响沸石的酸度,从而影响沸石酸性中心的静电场,并改变分子筛的催化性质。
2)裂化催化剂钒中毒机理。钒对沸石的中毒主要表现在使沸石结晶度下降,比表面积和孔体积减少,裂化活性降低。钒对沸石的破坏程度取决于钒的浓度、水热条件和沸石的类型。水在沸石钒中毒过程中起着重要作用,在水蒸汽存在的条件下,V2O5会发生以下反应:

为模拟工业催化剂在装置上受重全属镍钒影响的实际应用情况,按照所需污染量,将钒镍定量浸渍到催化剂上,将样品在120 ℃下烘干,然后在540 ℃下焙烧2 h即得污染催化剂样品,然后在800 ℃、100水蒸汽老化4 h时后测定污染样品的活性,结果见表15。
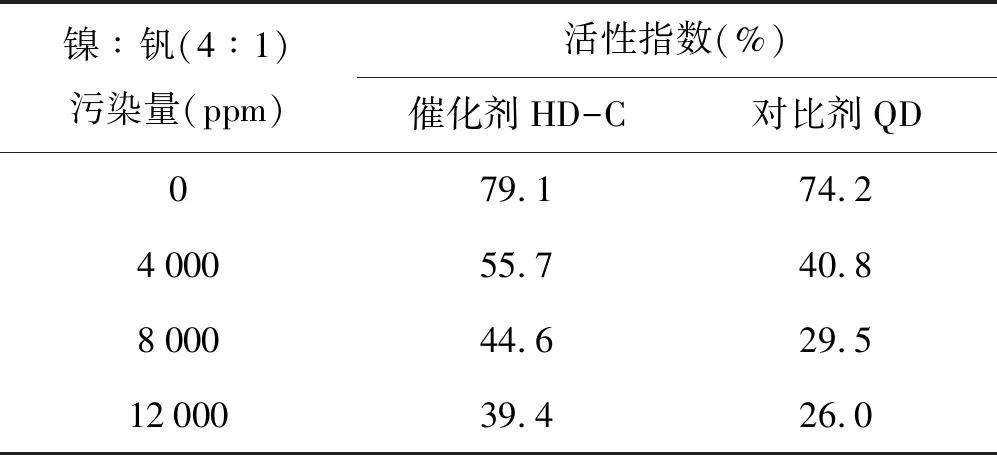
表15 催化剂不同钒镍污染量时的活性指数
注:800 ℃,100%水蒸汽老化4 h
在表15中列出了当钒镍污染量达到12 000 ppm时,催化剂HD-C微活指数为42.4%,对比剂QD仅为26%,说明催化剂HD-C具有较强的抗重金属钒镍污染能力。
2.2.2.6 催化剂的选择性

表16 催化剂的选择性
为了考察催化剂的选择性,将两种催化剂在800 ℃、100%水蒸汽老化4 h后测定其产品分布,原料使用减压渣油。结果见表16。
从表16可以看出:催化剂HD-C具有更高的转化率,可能老化时间短,导致两种催化剂的转化率都相对偏高;催化剂HD-C的干气偏高,这是不利的,其原因可能是由于催化剂老化时间又短,导致活性太高,产生过裂化所导致的;催化剂HD-C对汽油具有较好的选择性;其它特性如总液收、轻质油收率等催化剂HD-C都表现出一定的优势。
3 结论
笔者通过合成出NaY沸石晶化微球,通过铵根离子、稀土离子、磷酸二氢铵等进行改性、并通过两次焙烧,经由分析交换后母液来求得催化剂各成分含量后,进行催化剂性能评价,主要得到如下结论:
1)交换后催化剂上钠、稀土、铝、铁含量分别为0.28%、4.11%、36.24%、0.23%,总Na2O交换度达到96.0%,成品催化剂的Na2O含量达到工业催化剂的要求。
2)催化剂HD-C的结晶度为34%,基本满足商用催化剂对结晶度的要求;催化剂HD-C的比表面积和孔体积分别为400.2 m2·g-1和0.38 mL·g-1,均大于对比剂QD的31 m2·g-1和0.34 mL·g-1。从比表面和孔体积角度说明催化剂HD-C对反应物分子有更好的吸附、扩散和可接近性。
3)通过对催化剂HD-C和对比剂QD进行程序升温脱附发现:①催化剂HD-C在175 ℃和200 ℃处的吸附特性与QD相近;②两种催化剂300~400 ℃有较大差别,催化剂HD-C的吸附量大于对比剂QD;③催化剂HD-C的总算量(TPD图积分面积)大于对比剂QD。
4)催化剂理化性质和性能评价。通过常规化学分析方法、磨损指数仪及XRD分析得:HD-C和QD磨损指数及结晶度分别为1.1%·h-1、1.6%·h-1;34、28;可知HD-C结晶度较高、抗磨损性能较好;利用BET氮气吸附法考察了HD-C的比表面及孔体积得出催化剂HD-C的比表面积及孔体积大于对比剂QD,从比表面和孔体积角度说明催化剂HD-C对反应物分子有更好的吸附、扩散和可接近性;利用MAT考察了800 ℃、100%水蒸汽老化4 h、7 h、17 h和不同钒镍污染量的HD-C和QD的微反活性,得出催化剂HD-C与对比剂QD相比具有更高的催化活性、抗钒镍污染能力、选择性能优异。
猜你喜欢
辽宁化工(2022年8期)2022-08-27
岩性油气藏(2022年1期)2022-01-31
——水热过程影响机制
化工进展(2021年11期)2021-11-30
煤气与热力(2021年9期)2021-11-06
化学工业与工程(2021年5期)2021-11-03
石油炼制与化工(2021年9期)2021-09-15
建材发展导向(2021年13期)2021-07-28
湖南饲料(2021年3期)2021-07-28
丝绸(2020年6期)2020-06-23
江苏农业科学(2015年1期)2015-04-17
