
糖肾康胶囊的质量标准研究
2019-04-03 12:28黄涛
天津药学 2019年1期
黄 涛
(天津市儿童医院,天津 300134)
由于糖尿病肾病的病因病机还未搞清楚,因此西医针对这一疾病的治疗疗效总是不够理想,而中药复方具有多靶点、多途径和多层次的综合治疗优势,在临床上多有用于糖尿病肾病方面的治疗,疗效肯定[1]。因此,为满足临床应用需求,及时开发针对糖尿病肾病治疗的有效中药新药势在必行。糖肾康胶囊处方是来源于老中医临床经验方,由丹参、黄连、知母、水蛭、黄芪和淫羊藿等药材组成,临床应用时制备成汤剂,用于治疗糖尿病肾病,应用多年,疗效可靠。为了掩盖苦味、便于服用,考虑患者的顺应性,拟将其制成胶囊剂。其制备工艺为:黄连用乙醇单独提取,淫羊藿等6味药材采用水提醇沉工艺,分别制得干燥浸膏粉,混合后加辅料湿法制粒并装0#胶囊。本研究采用薄层色谱法(TLC)进行定性鉴别,分别鉴别了该制剂中的丹参、知母和黄芪;运用高效液相色谱法进行含量测定,选择的指标成分是处方中君药淫羊藿的主要化学成分淫羊藿苷,以为建立糖肾康胶囊的质量控制标准提供参考。
1 仪器与试药
DIONEX Ultimate型3000高效液相色谱仪(美国DIONEX公司);AUY220型万分之一电子分析天平(日本Shimadzu公司);AB135-S型十万分之一电子分析天平(瑞士Mettler-Toledo公司);BUG25-06型超声波清洗仪(上海必能信超声有限公司,功率:250 W,频率:25 kHz);Diamonsil C18色谱柱(200 mm×4.6 mm,5 μm,迪马科技有限公司);高效硅胶G板;乙腈、甲醇为色谱纯,其余试剂均为分析纯,水为超纯水。菝葜皂苷元对照品(批号110744-200509,纯度:95%)、丹酚酸B对照品(批号110532-201212,纯度>98%)、黄芪甲苷对照品(批号113791-200613,纯度>98%)、淫羊藿苷对照品(批号113717-200415,纯度>98%)、知母对照药材(批号120170-200503)、丹参对照药材(批号123823-201414)、黄芪对照药材(批号113681-200613)均购自中国食品药品检定研究院。实验所用药材均购自安国长安中药材有限公司,经天津药物研究院中药现代研究部张铁军研究员鉴定,符合《中国药典》2015年版一部要求。糖肾康胶囊(批号120803,120804,120805)为自制样品。各药味的阴性样品均由除该药味以外的其他药味按原处方、工艺制备。
2 方法与结果
2.1定性鉴别
2.1.1丹参 称取经研细的糖肾康胶囊内容物1.0 g,加10 ml无水乙醇,加稀盐酸调pH至2~3,超声(功率:250 W,频率:25 kHz)处理20 min,滤过,取续滤液作为供试品溶液。另取适量的丹酚酸B对照品,加甲醇溶解并定容,制备质量浓度为1 mg/ml的丹酚酸B对照品溶液。另取丹参对照药材过4号筛的细粉0.5 g,加25 ml甲醇,超声处理10 min,滤过,取续滤液作为对照药材溶液。按处方工艺和比例制备缺丹参的阴性样品,称取阴性样品1.0 g,按供试品溶液制备方法制成阴性对照溶液。按2015年版《中国药典》四部[2]TLC法试验,分别吸取以上4种溶液各10 μl,点到同一张硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲苯-甲醇-甲酸(3∶4∶2∶0.5∶2)为展开剂进行展开,取出薄层板,于通风橱中晾干,喷以3%FeCl3乙醇溶液显色,置日光灯下检视。结果在与对照品和对照药材薄层色谱的相应位置上,各供试品色谱显相同颜色的蓝色斑点,阴性样品色谱无干扰。见图1。

1.阴性对照 2.对照品 3.对照药材 4~6.供试品
2.1.2知母 称取经研细的本品内容物2.0 g,置于50 ml具塞锥形瓶中,加甲醇30 ml,超声处理30 min,过滤并蒸干滤液,残渣加水20 ml使溶解,溶液中加盐酸2 ml并摇匀;再加10 ml甲苯,水浴中加热回流提取1 h,自然冷却;分取甲苯层并蒸干,残渣加三氯甲烷1 ml使溶解,作为供试品溶液。另取适量的菝葜皂苷元对照品,加三氯甲烷溶解并定容,制备质量浓度为0.5 mg/ml的菝葜皂苷元对照品溶液。另取知母对照药材细粉(过4号筛)1.0 g,按供试品溶液制备方法制成对照药材溶液。按处方工艺和比例制备缺知母的阴性样品,称取阴性样品2.0 g,按供试品溶液制备方法制成阴性对照溶液。按2015年版《中国药典》四部[2]TLC法试验,分别吸取以上4种溶液各10 μl,点到同一张硅胶G薄层板上,以丙酮-环己烷-冰醋酸(1∶9∶0.2)为展开剂进行二次展开,取出薄层板,于通风橱中晾干,喷显色剂[8%香草醛无水乙醇-10%硫酸(1∶1)的混合溶液],于105 ℃烘至斑点显色清晰,置日光灯下检视。结果在与对照品和对照药材薄层色谱的相应位置上,各供试品色谱显相同颜色的黄色斑点,阴性样品色谱无干扰。见图2。
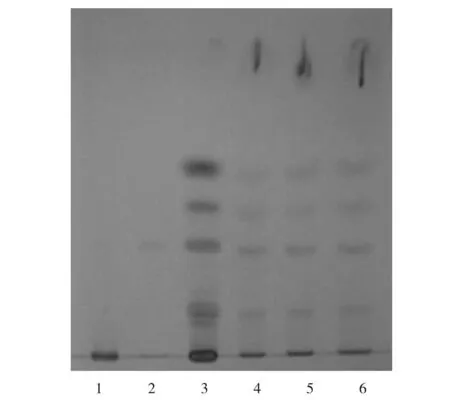
1.阴性对照 2.对照品 3.对照药材 4~6.供试品
2.1.3黄芪 称取经研细的本品内容物3.5 g,加30 ml甲醇,冷浸过夜,超声处理30 min,滤过,蒸干滤液,残渣加10 ml水并微热溶解,用正丁醇(水饱和)振摇萃取2次(每次30 ml),合并正丁醇液,用氨试液洗涤正丁醇液3次(每次20 ml),弃去氨液,蒸干正丁醇液,残渣加5 ml水使溶解;经大孔吸附树脂柱(12 cm×1.5 cm),按步骤洗脱:50 ml水→30 ml 40%乙醇溶液→60 ml 70%乙醇溶液,收集70%乙醇溶液洗脱液并蒸干,残渣加2 ml甲醇溶解,作为供试品溶液[3,4]。另取适量的黄芪甲苷对照品,加甲醇溶解并定容,制备质量浓度为1 mg/ml的黄芪甲苷对照品溶液。另取黄芪对照药材细粉(过4号筛)3.0 g,按供试品溶液制备方法制成对照药材溶液。按处方工艺和比例制备缺黄芪的阴性样品,称取阴性样品3.5 g,按供试品溶液制备方法制成阴性对照溶液。按2015年版《中国药典》四部[2]TLC法试验,分别吸取以上4种溶液各10 μl,点到同一张硅胶G薄层板上,以乙酸乙酯-氯仿-甲酸-水(20∶10∶11∶5)的下层溶液(混合液于10 ℃以下放置时分层)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇液,在105 ℃加热至斑点显色清晰,置日光灯下检视。结果,在与对照品和对照药材薄层色谱的相应位置上,各供试品色谱显相同颜色的棕褐色斑点,阴性样品色谱无干扰。见图3。

1.阴性对照 2.对照品 3.对照药材 4~6.供试品
2.2含量测定
2.2.1色谱条件 色谱柱:Diamonsil C18(200 mm×4.6 mm,5 μm);流动相为乙腈-水(27∶73);流速为1.0 ml/min;检测波长为270 nm;柱温为30 ℃;进样量为10 μl。
2.2.2溶液的制备
2.2.2.1对照品溶液的制备 精密称取适量的淫羊藿苷对照品,加甲醇溶解并定容,制成每1 ml含23.2 μg的溶液。
2.2.2.2供试品溶液的制备 精密称定经研细的本品内容物0.3 g,置于50 ml具塞锥形瓶中,精密加入20 ml甲醇,称定重量,超声处理30 min,自然冷却,再次称定重量,用甲醇补足减失的重量,摇匀,滤过,即得。
2.2.2.3阴性对照溶液的制备 按处方工艺和比例制备缺淫羊藿的阴性样品,精密称取阴性样品0.3 g,并按“2.2.2.2”项下方法制备淫羊藿阴性对照溶液。
2.2.3系统适用性试验 精密量取“2.2.2”项下对照品溶液、供试品溶液和阴性对照溶液各适量,按“2.2.1”项下色谱条件进样测定,记录色谱,详见图 4。由图4可知,在该色谱条件下进样分析,淫羊藿苷与其他成分可达到基线分离,分离度>1.5,理论塔板数不低于3 000。结果表明,其他成分对测定无干扰。
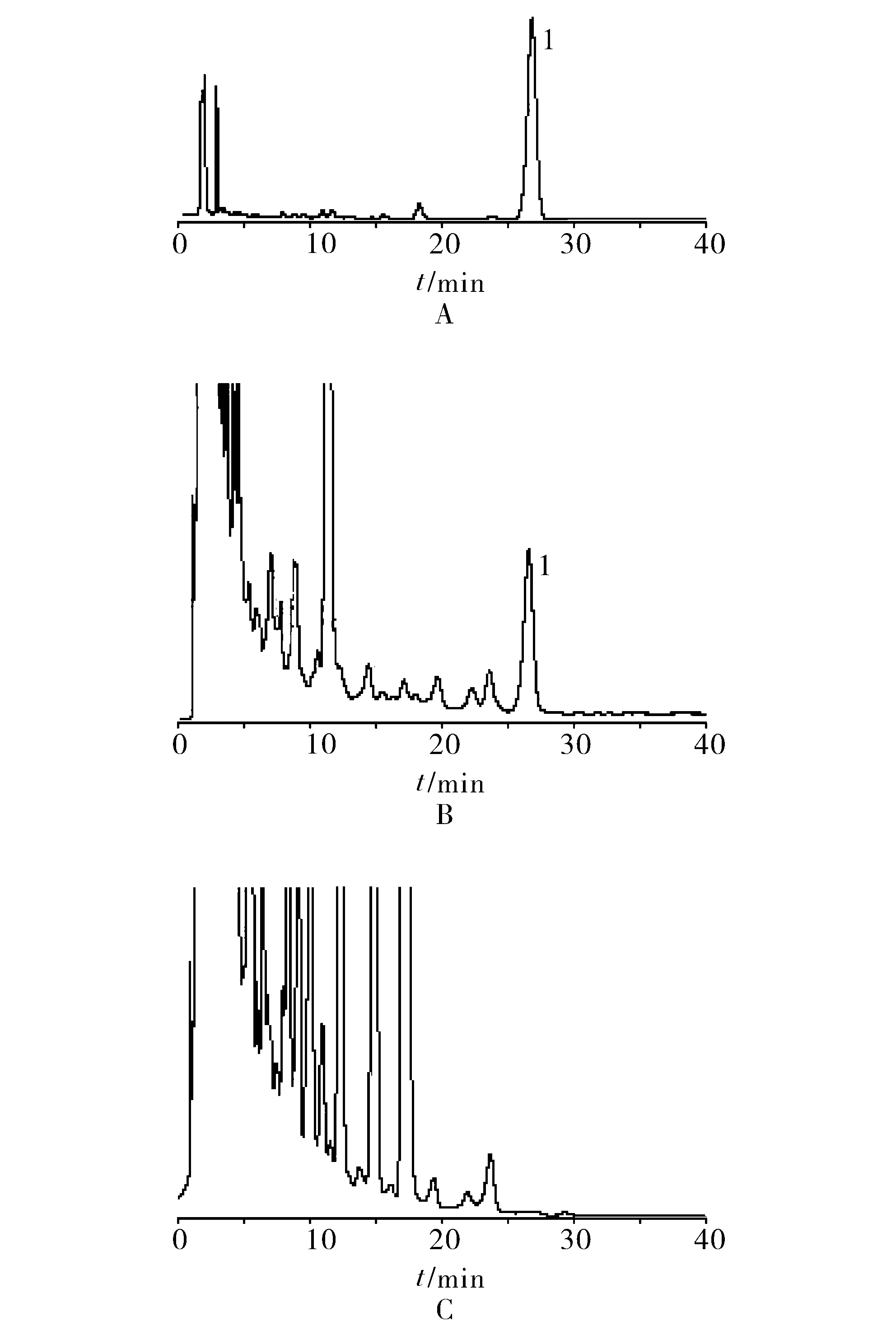
1.淫羊藿苷
2.2.4线性关系考查 精密吸取4、6、8、10、15和20 μl淫羊藿苷对照品溶液,按上述色谱条件进样分析。以进样量(X,μg)为横坐标、色谱峰面积(Y)为纵坐标进行线性回归,得回归方程为Y=2 479X-2.553 1(r=0.999 9)。结果表明,淫羊藿苷进样量在0.093~0.464 μg之间具有良好的线性关系。
2.2.5精密度试验 取适量的淫羊藿苷对照品溶液,按上述色谱条件连续进样分析6次,记录色谱峰面积。结果,淫羊藿苷峰面积RSD为0.93%(n=6),表明仪器精密度良好。
2.2.6稳定性试验 取适量的上述供试品溶液(批号120803),在室温下放置0、4、8、10和12 h时,再按上述色谱条件进样分析,记录色谱峰面积。结果,淫羊藿苷峰面积RSD为1.47%(n=5),表明供试品溶液在室温下放置12 h内基本稳定。
2.2.7重复性试验 精密称取适量的同一批号样品(批号120803),按“2.2.2.2”项下方法制备供试品溶液,共6份,再按上述色谱条件进样分析并计算含量。结果,淫羊藿苷的平均含量为1.403 1 mg/g,RSD为1.27%(n=6),表明本方法重复性良好。
2.2.8加样回收率试验 取适量的已知含量样品(批号120803),共9份,分别加入低、中、高质量的淫羊藿苷对照品,按“2.2.2.2”项下方法制备供试品溶液,再按上述色谱条件进样分析,记录色谱峰面积并计算加样回收率,结果平均加样回收率为99.18%,RSD为1.20%。见表1。
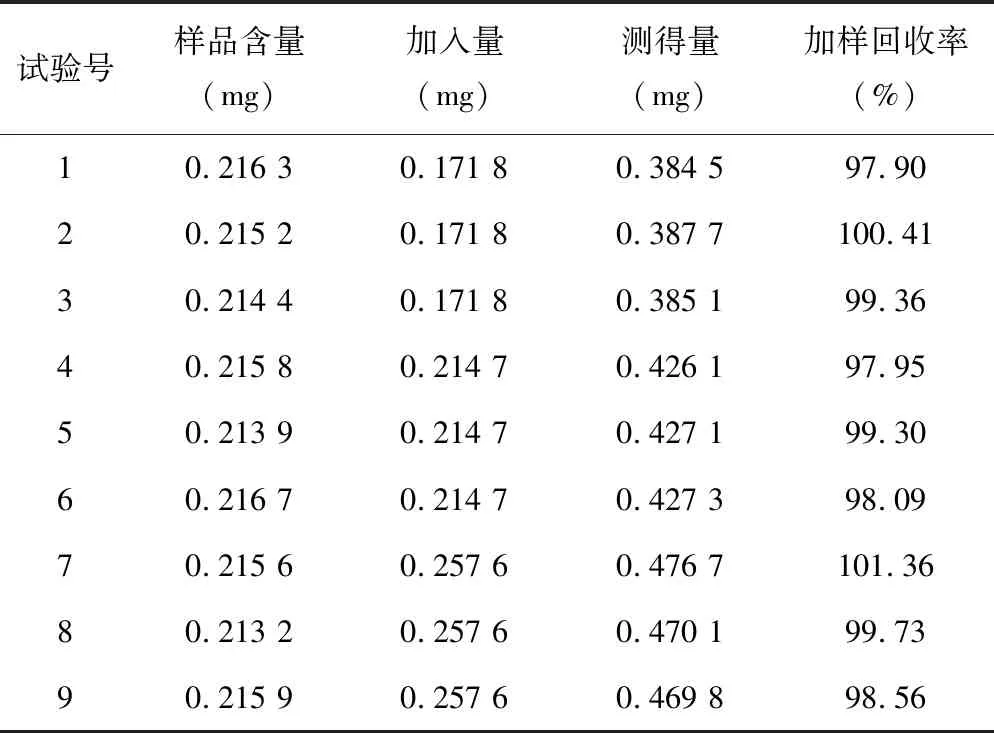
表1 淫羊藿苷加样回收率试验结果(n=9)
2.2.9样品中淫羊藿苷含量测定 每批取3份,分别按“2.2.2.2”项下方法制备供试品溶液,再按上述色谱条件进样分析,记录色谱峰面积并计算样品中淫羊霍苷含量,结果见表2。
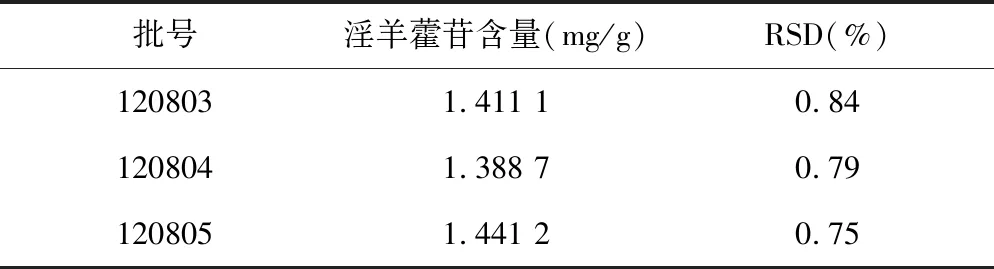
表2 样品含量测定结果(n=3)
3 结论与讨论
3.1定性鉴别 丹参、知母和黄芪为本方中主药,因而对这3味药进行定性鉴别,在制定鉴别方法时,从供试品溶液制备方法的选择、展开剂组成及比例的选择和拖尾控制等方面,参考了相关文献报道及《中国药典》方法,并进行了考查和优化,从而确定了最佳条件,方法简单可行。丹参薄层鉴别过程中,在处理方法选择时,分别考查了热回流和超声处理两种方法,最后选择采用超声处理的方法;在对丹参展开剂考查时,发现使用石油醚-乙酸乙酯-二氯甲烷为展开剂时阴性对照溶液有干扰斑点出现,故通过考查展开剂组成和比例,最终确定以三氯甲烷-乙酸乙酯-甲苯-甲醇-甲酸(3∶4∶2∶0.5∶2)为展开剂进行展开,结果分离效果良好且斑点清晰[5,6]。知母薄层鉴别过程中,参照文献[7,8]制备供试品溶液和选择展开剂,对加入盐酸的用量和时间进行了考查,结果表明,在刚开始直接加入盐酸水解,发现滤液黏稠,不容易过滤,等提取液滤过后再加入盐酸效果更好;分别加入盐酸1、2和3 ml进行试验,结果表明2 ml的量就符合要求。黄芪薄层鉴别过程中,参照文献[3]制备供试品溶液,对大孔吸附树脂因素和氨水洗涤因素进行了考查,最终确定利用氨试液洗涤,采用D101型大孔吸附树脂柱,试验结果较满意。
3.2含量测定 本研究采用高效液相色谱法,参考了2015年版《中国药典》一部[9]和相关文献报道[10-12],并进行了考查和优化,从而确定了最佳条件;提取溶剂考查时选择了50%乙醇、50%甲醇和甲醇,结果表明甲醇的提取效果最好;研究中还对超声提取时间进行了考查,分别超声提取20、30和40 min,结果表明提取30 min时样品中的淫羊藿苷已提取完全,且杂质较少。
综上所述,本方法简便、准确、灵敏度高,可用于糖肾康胶囊的质量控制。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
天津中医药大学学报(2022年4期)2022-08-27
青岛大学学报(医学版)(2022年3期)2022-08-05
现代中药研究与实践(2022年1期)2022-03-19
健康之家(2021年19期)2021-05-23
中国药学药品知识仓库(2021年18期)2021-02-28
国际放射医学核医学杂志(2021年10期)2021-02-28
中国骨质疏松杂志(2019年1期)2019-01-06
中国科技纵横(2018年2期)2018-11-29
海峡科技与产业(2016年3期)2016-05-17
