
DeepCPI:A Deep Learning-based Framework for Large-scale in silico Drug Screening
2019-03-07 07:27:24FangpingWanYueZhuHailinHuAntaoDaiXiaoqingCaiLigongChenHaipengGongTianXiaDehuaYangMingWeiWangJianyangZeng
Fangping Wan ,Yue Zhu ,Hailin Hu ,Antao Dai,Xiaoqing Cai,Ligong Chen ,Haipeng Gong ,Tian Xia ,Dehua Yang *,Ming-Wei Wang 7,8,*,Jianyang Zeng *
1 Institute for Interdisciplinary Information Sciences,Tsinghua University,Beijing 100084,China
2 The National Center for Drug Screening and the CASKey Laboratory of Receptor Research,Shanghai Instituteof Materia Medica,Chinese Academy of Sciences,Shanghai 201203,China
3 School of Medicine,Tsinghua University,Beijing 100084,China
4 School of Pharmaceutical Sciences,Tsinghua University,Beijing 100084,China
5 School of Life Science,Tsinghua University,Beijing 100084,China
6 Department of Electronics and Information Engineering,Huazhong University of Science and Technology,Wuhan 430074,China
7 School of Life Science and Technology,ShanghaiTech University,Shanghai 201210,China
8 Shanghai Medical College,Fudan University,Shanghai 200032,China
9 MOE Key Laboratory of Bioinformatics,Tsinghua University,Beijing 100084,China
Abstract Accurate identification of compound—protein interactions(CPIs)in silico may deepen our understanding of theunderlying mechanismsof drug action and thusremarkably facilitatedrug discovery and development.Conventional similarity-or docking-based computational methods for predicting CPIs rarely exploit latent features from currently available large-scale unlabeled compound and protein data and often limit their usage to relatively small-scale datasets.In the present study,we propose DeepCPI,a novel general and scalable computational framework that combines effective feature embedding(a technique of representation learning)with powerful deep learning methods to accurately predict CPIs at a large scale.DeepCPI automatically learns the implicit yet expressivelow-dimensional featuresof compoundsand proteinsfrom a massiveamount of unlabeled data.Evaluations of the measured CPIs in large-scale databases,such as ChEMBL and BindingDB,as well as of the known drug—target interactions from DrugBank,demonstrated the superior predictive performance of DeepCPI.Furthermore,several interactions among smallmolecule compounds and three G protein-coupled receptor targets(glucagon-like peptide-1 receptor,glucagon receptor,and vasoactive intestinal peptide receptor)predicted using DeepCPI were experimentally validated.The present study suggests that DeepCPI is a useful and powerful tool for drug discovery and repositioning.The source code of DeepCPI can be downloaded from https://github.com/FangpingWan/DeepCPI.
KEYWORDS Deep learning;Machine learning;Drug discovery;In silico drug screening;Compound—protein interaction prediction
Introduction
Identification of compound—protein interactions(CPIs;or drug—target interactions,DTIs)is crucial for drug discovery and development and provides valuable insights into the understanding of drug actions and off-target adverse events[1,2].Inspired by the concept of polypharmacology,i.e.,a singledrug may interact with multipletargets[3],drug developers are actively seeking novel ways to better characterize CPIs or identify novel uses of the existing drugs(i.e.,drug repositioning or drug repurposing)[3,4]to markedly reduce the time and cost required for drug development[5].
Numerous computational methods have been proposed to predict potential CPIs in silico to narrow thelargesearch space of possible interacting compound—protein pairs and facilitate drug discovery and development[6—12].Although successful results can be obtained using the existing prediction approaches,several challengesremain unaddressed.First,most of the conventional prediction methods only employ a simple and direct representation of features from the labeled data(e.g.,established CPIs and available protein structure information)to assess similarities among compounds and proteins and infer unknown CPIs.For instance,a kernel describing thesimilarities among drug—protein interaction prof iles[8]and the graph-based method SIMCOMP[13]wereused to comparedifferent drugs and compounds.In addition,the normalized Smith—Waterman score[9]istypically applied to assessthesimilaritiesamong targets(proteins).Meanwhile,largeamountsof availableunlabeled data of compounds and proteinsenablean implicit and useful representation of features that may effectively beused to definetheir similarities.Such an implicit representation of protein or compound features encoded by largescaleunlabeled data isnot exploited well by most of theexisting methodsto predict new CPIs.Second,an increasing number of established DTIsor compound—protein-binding aff inities(e.g.,1 million bioassays over 2 million compounds and 10,000 protein targets in PubChem[14])raises serious scalability issues concerning theconventional prediction methods.For instance,many similarity-based methods[7,9]require the computation of pairwise similarity scores between proteins,which is generally impractical in thesetting of large-scaledata.Theaforementioned computational challenges highlight the need of more efficient schemes to accurately capture the hidden features of proteins and compounds from massive unlabeled data as well astheneed of moreadvanced and scalablelearning modelsthat enable predictions from large-scale training datasets.
In machine learning communities,representation learning and deep learning(DL)are the two popular methods at present for efficiently extracting features and addressing the scalability issues in large-scale data analyses.Representation learning aims to automatically learn data representations(features)from relatively raw data that can bemoreeffectively and easily exploited by the downstream machine learning models to improve the learning performance[15,16].Meanwhile,DL aimsto extract high-level featureabstractions from input data,typically using several layers of non-linear transformations,and is a dominant method used in numerous complex learning tasks with large-scale samples in the data science field,such as computer vision,speech recognition,natural language processing (NLP),game playing,and bioinformatics [17—19].Although several DL models have been used to address various learning problems in drug discovery[20—22],they rarely fully exploit the currently available large-scale protein and compound data to predict CPI.For example,the computational approaches proposed in the literature[20,21]only use thehand-designed featuresof compoundsand do not takeinto account thefeaturesof targets.Furthermore,theseapproaches generally fail to predict potential interacting compounds for a given novel target(i.e.,without known interacting compounds in the training data);this type of prediction is generally more urgent than theprediction of novel compounds for targetswith known interacting compounds.Although a new approach—AtomNet—has been developed[23]to overcome these limitations,it can only be used to predict interacting drug partners of targets with known structures,which is often not the case in the clinical practice.In addition,despite the promising predictive performance of conventional approaches reported on benchmark datasets[24—26],few efforts have been made to explore the extent to which these advanced learning techniques can promote the efficiency in the real drug discovery scenario.
In this article,we propose Deep CPI,a novel framework that combines unsupervised representation learning with powerful DL techniques for predicting structure-free CPIs.DeepCPI first uses the latent semantic analysis[27]and Word2vec[16,28,29]methods to learn the feature embeddings(i.e.,low-dimensional feature representations)of compounds and proteins in an unsupervised manner from large compound and protein corpora,respectively.Subsequently,given a compound—protein pair,the feature embeddings of both compound and protein are fed into a multimodal deep neural network(DNN)classifier to predict their interaction probability.Wetested Deep CPI on several benchmark datasets,including the large-scale compound—protein aff inity databases(e.g.,ChEMBL and BindingDB),as well as the known DTIs from DrugBank.Comparisons with several conventional methods demonstrated the superior performance of Deep CPI in numerous practical scenarios.Moreover,starting from the virtual screening initialized by Deep CPI,we identified several novel interactions of small-molecule compounds with various targets in the G protein-coupled receptor(GPCR)family,including glucagon-like peptide-1 receptor(GLP-1R),glucagon receptor(GCGR),and vasoactive intestinal peptide receptor(VIPR).Collectively,our computational test and laboratory experimentation results demonstrate that Deep CPI is a useful and powerful tool for the prediction of novel CPIs and can thus aid in drug discovery and repositioning endeavors.
Results
DeepCPI framework
The Deep CPI framework comprises two main steps(Figure 1):(1)representation learning for both compounds and proteins and(2)predicting CPIs(or DTIs)through a multimodal DNN.More specifically,in the first step,we use several NLP techniques to extract the useful features of compounds and proteins from the corresponding large-scale unlabeled corpora(Figures S1 and S2;Materials and methods).Here,compounds and their basic structures are regarded as‘‘documents”and‘‘words”,respectively,whereas protein sequencesand all possible three non-overlapping amino acid residues are regarded as‘‘sentences”and‘‘words,”respectively.Subsequently,the feature embedding techniques,including latent semantic analysis[27]and Word2vec[16,28],are applied to automatically learn the implicit yet expressive low-dimensional representations(i.e.,vectors)of compound and protein features from the corresponding large-scale unlabeled corpora.In the second step,the derived low-dimensional feature vectors of compounds and proteinsare fed into a multimodal DNN classifier to make the predictions.Further details of the individual modules of DeepCPI,including the extraction of compound and protein features,DNN model,and implementation procedure,are described in Materials and methods.
Predictive performance evaluation
We mainly evaluated DeepCPI using compound—protein pairs extracted from the currently available databases,such as ChEMBL[30]and BindingDB[31].We first used the bioactivity data retrieved from ChEMBL[30]to assess the predictive performance of DeepCPI.Specifically,the compound—protein pairs with half maximal inhibitory concentrations(IC50)or inhibition constants(Ki)≤1μM were selected as positive examples,whereas pairs with IC50or Ki≥30μM were used as negative examples.This data preprocessing step yielded 360,867 positive examples and 93,925 negative examples.To justify our criteria of selecting positive and negative examples,we mapped the known interacting drug—target pairs extracted from DrugBank[32](released on November 11,2015)to the corresponding compound—protein pairs in ChEMBL(Materials and methods).The binding aff inities or potencies(measured by IC50or Ki)of majority of the known interacting drug—target pairs were≤1μM(>60%and 70%pairs for IC50and Ki,respectively)(Figure S3).Reportedly,1μM is a widely-used and good indicator of strong binding aff inities among compounds and proteins[33].Therefore,we considered IC50or Ki≤1μM as a reasonable criterion for selecting positive examples.There is no well-defined dichotomy between high and low binding aff inities;thus,we used a threshold of≥30μM(i.e.,markedly higher than 1μM)to select negative examples,which is consistent with the method reported elsewhere[23].
To evaluate the predictive performance of Deep CPI,we considered several challenging and realistic scenarios.A computational experiment was first conducted in which we randomly selected 20%pairs from ChEMBL as the training data and the remaining pairs as the test data.This scenario mimicked a practical situation in which CPIs are relatively sparsely labeled.ChEMBL may contain similar(redundant)proteins and compounds,which may lead to over-optimistic performance resulting from easy predictions.Therefore,we minimized this effect by only retaining proteins whose sequence identity scores were<0.40 and compounds whose chemical structure similarity scores were<0.55(as computed based on the Jaccard similarity between their Morgan fingerprints).More specifically,for each group of proteins or compounds with sequence identity scores≥0.40 or chemical structure similarity scores≥0.55,we only retained the protein or compound having the highest number of interactions and discarded the rest of the proteins or compoundsin that group.The basic statistics of the ChEMBL and BindingDB datasets used in our performance evaluation are summarized in Tables S1 and S2,respectively.
Conventional cross validation,particularly leave-one-out cross validation(LOOCV),may not be an appropriate method to evaluate the performance of a CPI prediction algorithm,if the training data contain many compounds or proteins with only one interaction[34].In such a case,training methods may learn to exploit thebiastoward theproteinsor compounds with a single interaction to boost the performance of LOOCV.Thus,separating the single interaction from other types of interactions during cross validation is essential[34].Given a compound—protein interacting pair from a dataset,if the compound or protein only appeared in this interaction,we considered this pair as unique(Materials and methods).In this test scenario,we used non-unique examples as the training data and tested the predictive performance on unique pairs.
In all computational tests,threebaselinemethodswereused for comparisons(Materials and methods).The first two were a random forest and a single-layer neural network(SLNN)with our feature extraction schemes.These were used to demonstrate the need for the DNN model.The third one was a DNN with conventional features(i.e.,Morgan fingerprints[35]with a radius of three for compounds and pairwise Smith—Waterman scores for proteins in the training data),which was used to demonstrate the need for our feature embedding methods.Moreover,we compared Deep CPI with two state-of-the-art network-based DTI prediction methods—DTINet[12]and NetLap RLS[10](Materials and methods)—in a setting where redundant proteins and compounds were removed;these two methods were not used in other scenarios(Figure S4)as the cubic time and quadratic space complexities concerning the large number of compounds exceeded the limit of our server.Weobserved that Deep CPI significantly outperformed the network-based methods(Figure 2A—D).Compared to these two network-based methods,DeepCPI achieved better time and space complexities(Materials and methods),demonstrating its superiority over network-based frameworks when handling large-scale data.In addition,DeepCPI outperformed the other three baseline methods(Figure 2A—D and Figure S4)and exhibited a better prediction accuracy and generalization ability of the combination of DL and our feature extraction schemes.
Furthermore,we conducted two supplementary tests to assess the predictive performance of DeepCPI on BindingDB(Tables S1 and S2).BindingDB stores the binding aff inities of proteins and drug-like small molecules[31]using the same criteria(i.e.,IC50or Ki≤1μM for positive examples and≥30μM for negative examples)to label compound—protein pairs.The compound—protein pairs derived from ChEMBL and BindingDB were employed as the training and test data,respectively.Compound—protein pairs from BindingDB exhibiting a compound chemical structure similarity score of≥0.55 and a protein sequence identity score of≥0.40 compared with any compound—protein pair from ChEMBL were regarded asoverlapsand removed from thetest data.Theevaluation results on the BindingDB dataset demonstrated that Deep CPI outperformed all of the baseline methods(Figure 2E and F;Figure S4).Collectively,these data support the strong generalization ability of DeepCPI.
We subsequently investigated the extraction of high-level feature abstractions from the input data using the DNN.We applied T-distributed stochastic neighbor embedding(t-SNE)[36]to visualize and compare the distributions of positive and negative examples with their original 300-dimensional input features and the latent features represented by the last hidden layer in DNN.In this study,DNN was trained on ChEMBL,and a combination of 5000 positive and 5000 negative examples randomly selected from BindingDB was used as the test data.Visualization(Figure S5)showed that the test data were better organized using DNN.Consequently,the final output layer(which was simply a logistic regression classifier)can more easily exploit hidden features to yield better classification results.
Finally,we compared the performance of DeepCPI with those of the following two DL-based models:AtomNet(a structure-based DL approach for predicting compound—protein binding potencies)[23]and Deep DTI[24](a deep belief network-based model with molecule fingerprints and protein k-mer frequencies as input features)(Materials and methods).Specifically,we compared Deep CPI with AtomNet in terms of the directory of useful decoys from DUD-E[37].DUD-E is a widely used benchmark dataset for evaluating molecular docking programs and contains active compounds against 102 targets(Table S3).Each active compound in DUD-E is also paired with several decoys that share similar physicochemical properties but have dissimilar two-dimensional topologies,under the assumption that such dissimilarity in the compound structure results in different pharmacological activities with high probability.

Figure 2 Performance evaluation of DeepCPI
We adopted two test settings as reported previously[23]to evaluate the performance of different prediction approaches using DUD-E.In the first setting,cross validation was performed on 102 proteins,i.e.,thedata wereseparated according to proteins.In the second setting,cross validation was performed for all pairs,i.e.,all compound—protein pairs were divided into three groups for validation.In addition to Atom-Net,we also compared our method with a random forest model.
The tests on DUD-E showed that DeepCPI outperformed both the random forest model and AtomNet in the aforementioned settings(Table S4).In addition,in the second setting,our protein structure-free feature extraction schemes with the random forest model also greatly outperformed AtomNet,which requires protein structures and uses a convolutional neural network classifier.These observations further demonstrate the superiority of our feature extraction schemes.DeepCPI achieved a significantly larger area under receiver operating characteristic curve(AUROC)than therandom forest model in the first test setting.The first setting was generally more stringent than the second one as protein information was not visible to classifiers during cross validation.Thus,this result indicates that Deep CPI has better generalization ability than the random forest model.
Deep DTI[24]requires high-dimensional features(14,564 features)as the input data;therefore,it can only be used for analyzing small-scale datasets.Thus,we mainly compared DeepCPI with Deep DTI using the 6262 DTIs provided by the original Deep DTI article[24].We applied the same evaluation strategy as that applied in Deep DTI by randomly sampling the same number(6262)of unknown DTIs as negative examplesand splitting the data into thetraining(60%),validation(20%),and test(20%)data.Our comparison showed that even on thissmall-scaledataset,Deep CPIcontinued to achieve a larger mean AUROC(0.9220)than Deep DTI(0.9158)(Table S5).Therefore,we believe that DeepCPI is superior to DeepDTI in terms of both predictive performance and scalability to large-scale compound aff inity data.
Novel interaction prediction
All known DTI data obtained from DrugBank[32](released on November 11,2015)wereused to train Deep CPI.Thenovel prediction results on the missing interactions(i.e.,the drug—target pairs that did not have an established interaction record in DrugBank)werethen examined.Most of thetop predictions with the highest scores could be supported by the evidence available in the literature.For example,among the list of the top 100 predictions,71 novel DTIs were consistent with those reported in previous studies(Table S6).Figure 3 presents the visualization of a DTI network comprising the top 200 predictions using Deep CPI as well as the network of the 71 aforementioned novel DTIs.
We describe several examples of these novel predictions supported by the literature below(Table S6).Specifically,in addition to the known DTIs recorded in DrugBank,DeepCPI revealed several novel interactions in neural pharmacology.These interactions may provide new direction to further decipher the complex biological processes in the treatment of neural disorders.For instance,dopamine,a catecholamine neurotransmitter with a high binding aff inity for dopamine receptors(DRs),was predicted by Deep CPI to also interact with theα2 adrenergic receptor(ADRA2A).Such a prediction representing a crosstalk within the evolutionally related catecholaminergic systems is supported by the known function of dopamine acting as a weak agonist of ADRA2A[38]as well as the evidence from multiple previous animal studies[39,40].Besides the intrinsic neurotransmitters,our prediction results involved various interesting interactions between other types of drugs and their novel binding partners.For instance,amitriptyline,a dual inhibitor of norepinephrine and serotonin reuptake,iscommonly used to treat major depression and anxiety.Our predictions indicated that amitriptyline can also interact with three DR isoforms,including DRD1,DRD2,and DRD3.This result is supported by previous evidence,suggesting that amitriptyline displays binding to all three DR isoforms at sub-micromolar potencies[41].
While these antagonist potencies are relatively weak compared with those of other targets(e.g.,solute carrier family 6 member 2[14]and histamine H1 receptor[42]),this new predicted interaction may offer expansion in the chemical space of antipsychotics[41]and the treatment of autism[43].Moreover,Deep CPI predicted that oxazepam,an intermediate-acting benzodiazepine widely used in the control of alcohol withdrawal symptoms,can also act on the translocator protein,an important factor involved in intromitochondrial cholesterol transfer.This prediction is also supported by previous data from radioligand binding assays[44]as well as by the observation that translocator protein is responsible for the oxazepam-induced reduction of methamphetamine in rats[45].
In addition to providing novel indications in neural pharmacology,our predictions showed that polythiazide,a commonly used diuretic,can act on carbonic anhydrases.This predicted interaction,which may be related to the antihypertensive function of polythiazide[38],is supported by the evidence that polythiazide serves as a carbonic anhydrase inhibitor in vivo[46].Another important branch of novel interaction predictions exemplif ied by an enzyme—substrate interaction between desipramine and cytochrome CYP2D6 highlighted a potential novel indication predicted by DeepCPI from a pharmacokinetics perspective.Indeed,the predicted interaction between desipramine and CYP2D6 is supported by their established metabolic association[47,48],thus offering important clinical implications in drug—drug interactions[49].Overall,the novel DTIs predicted by Deep CPI and supported by experimental or clinical evidence in the literature further demonstrate the strong predictive performance of Deep CPI.
Validation by experimentation
As 30%—40%of the marketed drugs target GPCRs[50,51],we applied Deep CPI to identify compoundsacting on this classof drug targets.In this experiment,we used positive and negative examples from ChEMBL and BindingDB as well as the compound—protein pairs with≤1μM aff inities in ZINC15[52—54]as the training data for DeepCPI.Brief ly,we predicted potential interacting compoundsusing a dataset obtained from the Chinese National Compound Library(CNCL;http://www.cncl.org.cn/,containing 758,723 small molecules)with three class B GPCRs(GLP-1R,GCGR,and VIPR)involved in metabolic disorders and hypertension[55,56].These proteins are challenging drug targets,particularly for the development of small-molecule modulators.For each GPCR target,we ran the trained Deep CPI model on the CNCL dataset and selected the top 100 predictions with the highest conf idence scores for experimental validation as detailed below.
Pilot screening
We first conducted several pilot screening assays as an initial experimental validation step to verify the top 100 compounds that were predicted by DeepCPI to act on the aforementioned three GPCRs.For GLP-1R,a whole-cell competitive binding assay was used to examine the effects of potential positive allosteric modulators(PAMs)(Figure4A;Materialsand methods).For GCGR and VIPR,a cAMPaccumulation assay was conducted to evaluate the agonistic and antagonistic activities of thepredicted compounds(Figure4B—E).A total of six putative ligands showed a significant augmentation of radiolabeled GLP-1 binding compared with DMSO control,i.e.,within the top 25%quantile of the maximum response(Figure 4A).Moreover,we discovered putative small-molecule ligands acting on GCGR and VIPR(Figure 4B—E).Among these,nine compounds exhibited significant antagonistic effects against GCGR(with 7%cAMP inhibition;Figure 4C),while one compound exhibited an obviousagonistic effect on VIPR(with 20%cAMP increase;Figure 4D).Thus,these hits were selected for further validation.
Conf irmation of PAMs of GLP-1R
The six putative hits were examined for their binding to GLP-1R.Of these,three(JK 0580-H009,CD3293-E005,and CD3848-F005;Figure S6)showed significant enhancement of GLP-1 binding to GLP-1R(Figure 5A).Their corresponding dose—response curves exhibited obvious positive allosteric effects,with half maximal effective concentration(EC50)values within the low micromolar range(<10μM;Figure 5B).To test the specificity of the three compounds,we investigated their binding ability to GCGR,a homolog of GLP-1R.These compounds did not cross-react with GCGR(Figure 5C)but substantially promoted intracellular cAMP accumulation in the presence of GLP-1(Figure 5D).Collectively,these results suggest that JK 0580-H009,CD3293-E005,and CD3848-F005 are PAMs of GLP-1R.

Figure 3 Network visualization of the novel DTIs predicted using DeepCPI
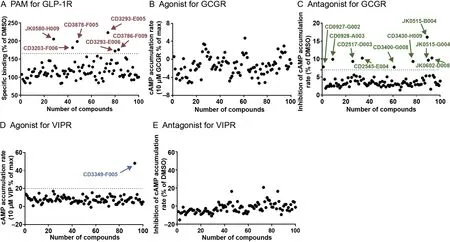
Figure 4 Pilot screening of the predicted compounds acting on GLP-1R,GCGR,and VIPR
To explore possible binding modes of these new PAMs,we conducted molecular docking studies using AutoDock Vina[57]based on the high-resolution three-dimensional active structure of GLP-1R[58](Figures 6 and Figure S6).We first used NNC0640(Figure S6),a negative allosteric modulator of GLP-1R[59],as a control to demonstrate that our docking approach could recover the experimentally solved complex structure(Figure S7).Interestingly,our docking results indicated that the binding pocket for the predicted PAMs are located between transmembrane helix 5(TM 5)and TM 6 of GLP-1R,which are distinct from that of NNC0640(Figure 6)and consistent with the enlarged cavity in the active form of GLP-1R(Figure S8).Additionally,the docking results suggest that the binding sites of our predicted PAMs are located deeper inside the transmembrane domain of GLP-1R than that of the known covalently bound PAMs,including Compound 2[59]and 4-(3-(benzyloxy)phenyl)-2-ethylsulf inyl-6-(trif luorome thyl)pyrimidine(BETP)[60,61](Figures 6A and S7).These findings reveal a novel route for discovering and designing new PAMs of GLP-1R.To further analyze these docking results,we produced four stable cell lines expressing mutant GLP-1Rs(C347F,T149A,T355A,and I328N).As a control,we first measured the activity of BETP,which is covalently bonded to C347 in GLP-1R.Consistent with the previous findings[59],T149A mutation diminished the binding between125I-GLP-1 and GLP-1R,which could be restored by BETP treatment(Figure 7).Meanwhile,C347F mutation eliminated the covalent anchor of BETP and reduced its eff icacy compared with that of wild-type GLP-1R(Figure 7).
However,none of the three predicted compounds exhibited binding to the T149A mutant,and their modulation behavior on C347F mutant generally aligned with that on wild-type,supporting its non-covalent binding nature(Figure 7,Table S7).These observations point to a divergent binding mode of the predicted PAMs different from that of BETP.Intriguingly,I328N mutation principally abolished the allosteric effects of thecompounds(Figure7),probably dueto a largesteric crash,aspredicted by thedocking study.In contrast,T355A mutation located at theother sideof TM6(Figure6)showed a negligible impact on the PAM activities of the predicted compounds(Figure 7 and Table S7).Collectively,our mutagenesis results support the docking data,indicating that DeepCPI can discover potential PAMs of GLP-1R.

Figure 5 Experimental validation of the putative PAMs of GLP-1R
Validation of GCGR and VIPR modulators
Nine hits with antagonistic effects against GCGR(Figure 4C)were identified in the pilot screening.Among these,CD3400-G008(Figure S6)was conf irmed to stably decrease glucagoninduced cAMP accumulation(Figure 8A).Subsequently,its dose dependency and estimated IC50value of antagonism(22.6μM)were determined(Figure 8B).In addition,this compound led to a rightward shift of the glucagon dose—response curve,as measured by the cAMP accumulation assay(Figure 8C).This shift corresponded to an increase in the EC50value of glucagon from 23.9 p M to 5.56 pM,although it did not affect forskolin-induced cAMPaccumulation(Figure 8D),ruling out the possibility that CD3400-G008 decreases cAMP accumulation in a non-specific manner.Similarly,theagonistic effect of the putative VIPR agonist CD3349-F005(Figures 4 and S6)was dose-dependent(Figure 8E),while its agonism specificity was conf irmed using a phosphodiesterase(PDE)inhibitor exclusion assay(Figure 8F).The results showed that neither 25μM nor 50μM of CD3349-F005 affected forskolininduced cAMP accumulation.
Collectively,these data support the notion that DeepCPI prediction can offer a promising starting point for smallmolecule drug discovery targeting GPCRs.
Discussion
In this article,we propose DeepCPI as a novel and scalable framework that combines data-driven representation learning with DL to predict novel CPIs(DTIs).By exploiting the large-scale unlabeled data of compounds and proteins,the employed representation learning schemes effectively extract low-dimensional expressive features from raw data without therequirement for information on protein structureor known interactions.
The combination of the effective feature embedding strategies and the powerful DL model is particularly useful for fully exploiting the massive amount of compound—protein binding data available from large-scale databases,such as PubChem and ChEMBL.The effectiveness of our method was fully validated using several large-scale compound—protein binding datasets as well as the known interactions between Food and Drug Administration(FDA)-approved drugs and targets.Moreover,we experimentally validated several compounds that were predicted to interact with GPCRs,which represent the largest transmembrane receptor family and probably the most important drug targets.This family constitutes>800 annotated and 150‘‘orphan”receptors.The latter are without known endogenous ligands and/or functions.Target-based drug discovery has been a focal point of research for decades.However,the inefficiency of mass random bioactivity screening promotes the application ofin silicoprediction and discovery of small-molecule ligands.Our DeepCPI model establishes a new framework that effectively combines feature embedding with DL for the prediction of CPIs at a large scale.

Figure 6 Molecular docking results of the predicted PAMs of GLP-1R
We conducted several functional assays to validate our prediction results regarding the identification of small-molecule modulators targeting several class B(i.e.,the secretin-like family)GPCRs.GLP-1R is an established drug target for type 2 diabetes and obesity,and several peptidic therapeutic agents have been developed and marketed with combined annual sales of billions of dollars.As most therapeutic peptides require non-oral administration routes,discovery of orally available small-molecule surrogates is highly desirable.To the best of our knowledge,since the discovery of Boc5—the first non-peptidic GLP-1R agonist—more than a decade ago[62—64],little progress has been made in identifying‘‘druggable”small-molecule mimetics for GLP-1.In this study,we identified three PAMs that were computationally predicted by Deep CPI and experimentally conf irmed with bioassays to be specific to GLP-1R,thereby providing an alternative to discover non-peptidic modulators of GLP-1R.
The docking results of our predicted hits demonstrated that they could be fitted to similar sites corresponding to the binding pockets for previously known PAMs at GLP-1R in its active form.The experimental data generated by binding and cAMP accumulation assays conf irmed the positive allosteric action of thesehits.Overall,our modeling data,in conjunction with those obtained from mutagenesis studies,revealed the binding poses of the predicted interactions between these compounds and GLP-1R.These results offer new insights into the structural basis and underlying mechanisms of drug action.
Cross validation through different databases,supporting evidence from the known DTIs in the literature,and laboratory experimentation indicate that DeepCPIcan serveasa useful tool for drug discovery and repositioning.In our follow-up studies,weintend to combine DeepCPIwith additional validation experiments for the discovery of drug leads against a wide range of targets.Better prediction results may be achieved by incorporating other available data,such as gene expression and protein structures,into our DL model.
Materials and methods
DeepCPI
Deep CPI is an extension of our previously developed CPIprediction model[65].We describe the building blocks of Deep CPI in the following three subsections.
Compound feature extraction

Figure 7 Mutations in GLP-1R affect the activities of predicted PAMs
To learn good embeddings(i.e.,low-dimensional feature representations)of compounds,we used the latent semantic analysis(also termed latent semantic indexing)technique[27],which is probably one of the most effective methods for document similarity analyses in the field of NLP.In latent semantic analysis,each document is represented by a vector storing the term frequency or term frequencyinverse document frequency information(tf-idf).This is a numerical statistic widely used in information retrieval to describe the importance of a word in a document.Subsequently,a corpus(i.e.,a collection of documents)can be represented by a matrix,in which each column stores the tf-idfscores ofindividual terms in a document.Subsequently,singular value decomposition(SVD)is applied to obtain low-dimensional representations of features in documents.

Figure 8 Experimental validation of the hit compounds acting on GCGR and VIPR
In the context of compound feature extraction(Figure S1),a compound and its substructures can be viewed as a document and corresponding terms,respectively.Given a compound set N,we use the Morgan fingerprints[35]with a radius of one to scan every atom of each compound in N and then generate the corresponding substructures.Let D denote the set of substructures generated from all compounds in N.We then employ a matrix M∈RD||×|N|to store the word count information for these compounds,where Mijrepresents the tf-idf value of the ithsubstructure in the jthcompound.More specifically,Mijis defined as tf (i,j )·idf ( i,N ),where tf ( i,j )stands for the number of occurrences of the ithsubstructure in the jthcompound,and idf( i ,N)=.Here,{j∈D:tf( i ,j)≠0}represents the number of documents containing the ithsubstructure.Basically,idf ( i,N)reweighs tf ( i,j ),resulting in lower weights for more common substructures and higher weights for less common substructures.This is consistent with an observation in the information theory that rarer events generally have higher entropy and are thus more informative.
After M is constructed,it is then decomposed by SVD into three matrices,U,Σ,V*,such that M=UΣV*.Here,Σis a|D|×|N|diagonal matrix with the eigenvalues of M on the diagonal matrix,and U isa| D|×|D|matrix in which each column Uiis an eigenvector of M corresponding to the itheigenvalueΣii.
To embed thecompoundsinto a low-dimensional space Rd,where d<D||,we select the first d columns of U,which correspond to the largest eigenvaluesinΣ.Letdenotethematrix with columns corresponding to these selected eigenvectors.Subsequently,a low-dimensional embedding of M can be obtained byM,whereisa d×|N|matrix,in which the ithcolumn corresponds to a d-dimensional embedding(or embedded feature vector)of the ithcompound.can be precomputed and fixed after being trained from a compound corpus.Given any new compound,its embedded low-dimensional feature vector can be obtained by left multiplying its tf-idf by(Figure S1).
Our compound feature embedding framework used the compounds retrieved from multiple sources,including all compounds labeled as active in bioassays on PubChem[14],all FDA-approved drugs in DrugBank[32],and all compounds stored in ChEMBL[30].Duplicate compounds were removed according to their International Chemical Identifiers(InChIs).Thetotal number of final compoundsin our compound feature extraction module used to construct matrix M was 1,795,801.The total number of different substructures generated from the Morgan fingerprints with a radius of one was 18,868.We setd=200,which is a recommended value in latent semantic analysis[66].
Protein feature extraction
We applied Word2vec,a successful word-embedding technique widely used in various NLP tasks[16,28],to learn the lowdimensional representations of protein features.In particular,we use the Skip-gram with a negative sampling method[28]to train the word-embedding model and learn the context relationsbetween wordsin sentences.Wefirst introducesome necessary notations.Supposethat we aregiven a set of sentencesSand a context window of sizeb.Given a sentences∈Sthat is represented by a sequence of words,whereLis the length ofs,the contexts of a wordwsiare defined asThat is,all the words appearing within the context window of sizeband centered at wordwin the sentence are regarded as the contexts ofw.We further useWto denote the set of all words appearing inS,and#(w,c)to denote the total number of occurrences of wordc∈Wappearing in the context window of the wordw∈WinS.Since each word can play two roles(i.e.,center word and context word),the Skip-gram method equips every wordw∈Wwith twod-dimensional vector representations ew,aw∈Rd,where ewis used whenwis a center word and awisused otherwise(both vectorsarerandomly initialized).Here,ewor awbasically represents the coordinates ofwin the lower dimensional(i.e.,d-dimensional)space after embedding.Subsequently,our goal is to maximize the following objective function:

One problem in this objective function(i.e.,Equation 1)is that it does not take into account any negative example.If we arbitrarily assign any large positive values to ewand aw,σ(ew·aw)would invariably be predicted as 1.In this case,although Equation 1 ismaximized,such embeddings are surely useless.To tackle this problem,a Skip-gram model with negative sampling[28]has been proposed,in which‘‘negative examples”cnscns( ∈Wandcns≠w)are drawn from a data distributionwhereMrepresents the total number of words inSand#cnsrepresents the total number of occurrences of wordcnsinS.Then,the new objective function can be written as follows:wherekis the number of‘‘negative examples”to be sampled for each observed word—context pairw,c( )during training.Maximizing this objective function can be performed using the stochastic gradient descent technique[16].

For each observed word—context pairw,c( ),Equation 2 aims to maximize its log-likelihood,while minimizing the log-likelihood ofkrandom pairs(w,cns)under the assumption that such random selections can well ref lect the unobserved word—context pairs(i.e.,negative examples)representing the background.In other words,the goal of this task is to distinguish the observed word—context pairs from the background distribution.
As in other existing schemes for encoding the features of genomic sequences[29],each protein sequence in our framework is regarded as a‘‘sentence”reading from its Nterminus to C-terminus and every three non-overlapping amino acid residues are viewed as a‘‘word”(Figure S2).For each protein sequence,we start from the first,second,and third amino acid residues from the N-terminus sequentially and then consider all possible‘‘words”while discarding those residues that cannot form a‘‘word”.
After converting protein sequences to‘‘sentences”and all three non-overlapping amino acid residues to‘‘words”,Skipgram with negative sampling is employed to learn the lowdimensional embeddings of these‘‘words”.Subsequently,the learnt embeddings of‘‘words”are fixed,and an embedding of a new protein sequence is obtained by summing and averaging theembeddings of all‘‘words”in all threepossible encoded‘‘sentences”(Figure S2).Of note,a similar approach has been successfully used to extract useful features for text classification using Word2vec[67].
In our study,the protein sequences used for learning the low-dimensional embeddings of protein features were retrieved from several databases,including PubChem[14],DrugBank[32],ChEMBL[30],Protein Data Bank[68](www.rcsb.org),and UniProt[38].All duplicate sequences were removed,and the final number of sequences for learning the protein features during the embedding process was 464,122.We followed the previously described principles[29]to select the hyper parameters of Skip-gram.More specifically,the embedding dimension was set tod=100,the size of the context window was set tob=12,and the number of negative examples was set tok=15.
Multimodal DNN
Suppose that we are given a training dataset of compound—protein pairs1,2,...,n} and a corresponding label set {yi|i=1,2,...,n},wherenstands for the total number of compound—protein pairs,yi=1 indicates that compoundciand proteinpiinteract with each other,andyi=0 otherwise.We first use the feature extraction schemes described earlier to derive the feature embeddings ofindividual compounds and proteins,and then feed these two embeddings to a multimodal DNN to determine whether the given compound—protein pair exhibits a true interaction.
We first introduce a vanilla DNN and then describe its multimodal variant.The basic DNN architecture comprises an input layerL0,an output layerLout,andHhidden layersLh(h∈ {1 ,2,...,H})between input and output layers.Each hidden layerLhcontains a set of units that can be represented by a vector ah∈R|Lh|,where|Lh|stands for the number of units inLh.Subsequently,each hidden layerLhcan be parameterized by a weight matrix Wha bias vector bh∈R|Lh|,and an activation functionf(·).More specifically, the units inLhcan be calculated bywhereh=1,2,...,H,and the units a0in the input layerL0are the input features.We use the rectified linear unit functionf(x)=max ( 0,x),which is a common choice of activation function in DL,perhaps due to its sparsity property,high computational efficiency,and absence of the gradient-vanishing effect during the back-propagation training process[69].
The multimodal DNN differs from the vanilla DNN in terms of the use of local hidden layers to distinguish different input modalities(Figure 1).In our case,the low-dimensional compound and protein embeddings are considered distinct input modalities and separately fed to two different local hidden layers.Subsequently,thesetwo types of local hidden layers are concatenated and fed to joint hidden layers(Figure 1).Here,the explicit partition of local hidden layers for distinct input channels can better exploit the statistical properties of different modalities[70].
After we calculate the aHfor the final(joint)hidden layer,the output layerLoutis simply a logistic regression model that takes aHas its input and computeswhere the output^yis the conf idence score for classification,σis the sigmoid function,wout∈R|LH|,andbout∈R are the parameters of the output layerLout.Since the sigmoid function has a range (0,1),can also be interpreted as the interacting probability of the given compound—protein pair.
To learn wout,bout,and all parameters Wh,bhin the hidden layers from the training data set and the corresponding label set,we need to minimize the following cross-entropy loss:

The aforementioned minimization problem can be solved using the stochastic gradient descent and back-propagation techniques[19].In addition,we apply two popular strategies in DL communities—dropout[71]and batch normalization [72]—to further enhance the classification performance of our DL model.In particular,dropout sets the hidden units to zero with a certain probability,which can effectively alleviate the potential overfitting problem in DL[71].The batch normalization scheme normalizes the outputs of hidden units to zero mean and unit standard deviation,which can accelerate the training process and act as a regularizer[72].
Since positive and negative examples are possibly imbalanced,our classifier may learn a‘‘lazy”solution.That is,in such a skewed data distribution case,the classifier can relatively easily predict the dominant class given any input.To alleviate this problem,we downsample the examples from the majority class in order for thenumbersof positiveand negative examples to be comparable during training.In our computational tests,we implement an ensemble version of the previously described DL model and use the average prediction over 20 models to obtain relatively more stable classification results.
Time and space complexities
Training
For compound feature extraction,given a compound,letgdenote the running time of generating its Morgan fingerprints with a radius of one.The time complexity for extracting the low-dimensional representations of compound features is
For the protein feature extraction process,given a protein corpusS,it takestime andspace to scan and calculate the context information,whereWandbstand for the set of all possible three non-overlapping amino acid residues and the context window size,respectively.Given a word—context pair(w,c)∈W×W,it takestime to compute the objective function in Equation 2,wheredpstands for the dimension of a protein embedding andkstands for the number of negative samples for each word—context pair.Suppose that we performqiterations of gradient descent,then,the time complexity of the protein feature extraction module isAfter training,we needspace to store the protein embeddings for future inference.
For a neural network,let|Lh-1|and|Lh|denote the numbers of units in layersh-1 andh,respectively.Suppose that|Lh-1|×|Lh|isthelargest valueamong all layers,then,the time and spacecomplexitiesfor training our DL model areboundedandrespectively,wherenstands for the total number of training samples,Hstands for the number of hidden layers in the neural network,andqstands for the number of training iterations.
Prediction
During the prediction stage,given a compound—protein pair,we first compute the low-dimensional vector representations of their features.This operation takestime for the compound andtime for the protein,whererstands for the length of the protein sequence.Then,these two lowdimensional vector representations are fed to the deep multimodal neural network to make the prediction,which takestime.In our framework,we set|Lh-1|=1024 and| |=256,which are small and can be considered constant(also see‘‘Implementation of Deep CPI”below).
Mapping DrugBank data to ChEMBL
A known drug—target pair from DrugBank[32]is considered to bein ChEMBL[30]if compound—target pairs with identical InChIs for compounds or drugs and sequence identity scorest≥0.40 for proteins are present in the latter dataset.Given two protein sequencessands’,their sequence identity scoretis defined ast=whereSW(· ,·)stands for the Smith—Waterman score[73].
Def inition of unique compounds and proteins
Given a dataset,a compound is considered unique if its chemical structure similarity score with any other compound is<0.55,where the chemical structure similarity score is defined asthe Jaccard similarity between two Morgan fingerprintswith a radius of three of the corresponding compounds[35].Similarly,a protein is considered unique if its sequence similarity score with any other protein is<0.40.
DeepCPI implementation
The Morgan fingerprints of compounds were generated by RDKit(https://github.com/rdkit/rdkit).Latent semantic analysis and Word2vec(Skip-gram with negative sampling)were performed using Gensim[74],a Python library designed to automatically extract semantic topics from documents.Our DNN implementation was based on Keras(https://github.com/keras-team/keras)—a highly modular DL library.For all computational experiments,weused two local hidden layers with 1024 and 256 units,respectively,for both compound and protein input channels.The two local layers were then connected to three joint hidden layers with 512,128,and 32 units,respectively.We set the dropout rate at 0.2.Batch normalization was added to all hidden layers.During training,we selected 20%of the training data as validation set to select the optimal training epoch.
Baseline methods
When testing on ChEMBL[30]and BindingDB[31],we compared our method with two network-based DTI prediction methods,including DTINet[12]and NetLap RLS[10],as well as with three constructed baseline methods as shown below.
For DTINet and NetLap RLS,Jaccard similarity between the Morgan fingerprintsof thecorresponding compoundswith a radius of three and pairwise Smith—Waterman scores were used to construct compound and protein similarity matrices as required in both methods.The default hyperparameters of both methods were used.
For random forest with our feature extraction schemes,we set the tree number to 128 in all computational tests as previously recommended[75].We randomly selected 20%of the training data as validation set to select the optimal tree depth from 1 to 30.
For SLNN with our feature extraction schemes,we used a local hidden layer with 1024 unitsfor both compound and protein input channels.The two local layers were then connected and fed to a logistic layer to make the CPI prediction.We set the dropout rate to 0.2 and batch normalization was added to the hidden layer as in Deep CPI.We selected 20%of the training data as validation set to select the optimal training epoch.
For DNN with conventional features,instead of using our feature extraction schemes,Morgan fingerprints with a radius of three and pairwise Smith—Waterman scores were used as compound and protein features,respectively.These features were subsequently fed into the same multimodal neural network as in Deep CPI.We selected 20%of the training data as validation set to select the optimal training epoch.
We also compared our method Deep CPI with two other DL-based models,namely AtomNet[23]and DeepDTI[24].
When comparing with AtomNet,we experienced diff iculty in reimplementing AtomNet.Therefore,we mainly compared the performance of Deep CPI with that of AtomNet on the same DUD-E dataset.For a fair comparison,we only used a single Deep CPI model instead of an ensemble version.
We also compared the performance of Deep CPI to that of DeepDTI on 6262 DTIs provided by the original DeepDTI article[24].DeepDTI conducted a grid search to determine the hyper parameters of the model.Hence,for a fair comparison,we followed the same strategy to determine the hyper parameters of Deep CPI.Here,we reported the hyper parameter space that we searched.In particular,we selected a batch size from {32,128,512},dimensions of compound and protein features from {50,60,70,80,90,100,200,300},dropout rate from{ 0 .1,0.2},and joint hidden layers with sizes from{{512,128,32},{5 12,64}}.We only used a single DeeCPI model instead of an ensemble version for a fair comparison.
Molecular docking
Compounds were docked using AutoDock Vina[57].The GLP-1R model in its active form was extracted from a cocrystal structure of full-length GLP-1R and a truncated peptide agonist(PDB:5NX2)[58].The best docked poses were selected based on the Vina-predicted energy values.
Experimental validation
Cell culture
Stable cell lines were established using FlpIn Chinese hamster ovary(CHO)cells(Invitrogen,Carlsbad,CA)expressing either GLP-1R or GCGR and cultured in Ham’s F12 nutrient medium(F12)with 10%fetal bovine serum(FBS)and 800μg/ml hygromycin-B at 37°C and 5%carbon dioxide(CO2).Desired mutationswereintroduced to GLP-1R construct using the Muta-directTMkit(Catalog No.SDM-15;Beijing SBSGenetech,Beijing,China)and integrated into FlpIn-CHO cells.VIPR overexpression was achieved through transient transfection using Lipofectamine 2000(Invitrogen)in F12 medium with 10%FBS.Cells were cultured for 24 h before being seeded into microtiter plates.
Whole-cell competitive ligand binding assay
CHO cells stably expressing GLP-1R or GCGR were seeded into 96-well plates at a density of 3×104cells/well and incubated overnight at 37°C and 5%CO2.The radioligand binding assay was performed 24 h thereafter.For homogeneous binding,cells were incubated in binding buffer with a constant concentration of125I-GLP-1(40 p M,PerkinElmer,Boston,MA)or125I-glucagon(40 p M,PerkinElmer)and unlabeled compounds at 4°C overnight.Cells were washed three times with ice-cold PBSand lysed using 50μl lysis buffer(PBSsupplemented with 20 mM Tris—HCl and 1%Triton X-100,p H 7.4).Subsequently,the plates were counted for radioactivity(counts per minute,CPM) in a scintillation counter(MicroBeta2 Plate Counter,PerkinElmer)using a scintillation cocktail(OptiPhase SuperMix;PerkinElmer).
cAMP accumulation assay
All cells were seeded into 384-well culture plates(4000 cells/well)and incubated for 24 h at 37°C and 5%CO2.For the agonist assay,after 24 h,the culture medium was discarded and 5μl cAMP stimulation buffer[calcium-and magnesiumfree Hanks’balanced salt solution(HBSS)buffer with 5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid(HEPES),0.1%bovine serum albumin(BSA),and 0.5 mM 3-isobutyl-1-methylxanthine(IBMX)]was added to the cells.Subsequently,5μl compounds were introduced to simulate the cAMP reaction.For the PAM or antagonist assay,each well contained 2.5μl cAMP stimulation buffer,5μl endogenous ligand(GLP-1,glucagon,or VIP)at various concentrations,and 2.5μl testing compounds diluted in the cAMP assay buffer.After 40-min incubation at room temperature,cAMP levels were determined using the LANCE cAMP kit(Catalog No.TRF0264;PerkinElmer).
Specificity verification and PDE inhibitor exclusion assay
Two experiments were performed using the cAMP accumulation assay(antagonist mode)to study the specificity of the hit compounds acting on GCGR or VIPR.In the case of GCGR,glucagon was replaced by forskolin to investigate whether CD3400-G008 affects forskolin-induced cAMP accumulation. Forskolin concentration was gradually increased from 1.28 nM to 100μM,whereas CD3400-G008 concentration was kept unchanged(20μM).The PDE inhibitor exclusion assay was performed in CHO-K 1 cells,in which IBMX-free stimulation buffer (calcium- and magnesium-free HBSS buffer with 5 mM HEPES and 0.1%BSA)was used.Concentrations of both IBMX(PDE inhibitor,positive control)and forskolin were gradually increased from 1.28 nM to 100μM,and the agonistic effect of CD3349-F005 was examined at concentrations of 25μM and 50μM,respectively.
Availability
The source code of Deep CPI can be downloaded from https://github.com/FangpingWan/Deep CPI.
Authors’contributions
FW,DY,MWW,and JZ conceived the project.JZ,DY,and MWW supervised the project.FW and JZ designed the computational pipeline.FW implemented DeepCPI,and performed the model training and prediction validation tasks.HH and JZ analyzed the novel prediction results.YZ,AD,XC,and DY performed the experimental validation.DY and MWW analyzed the validation results.HH and HG performed the computational docking and data analysis.TX and LC helped analyze the prediction results.FW,HH,MWW,and JZ wrote the manuscript with input from all co-authors.All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China(Grant Nos.61872216 and 81630103 to JZ,81872915 to MWW,81573479 and 81773792 to DY),the National Science and Technology Major Project(Grant No.2018ZX09711003-004-002 to LC),the National Science and Technology Major Project Key New Drug Creation and Manufacturing Program of China(Grant Nos.2018ZX09735-001 to MWW,2018ZX09711002-002-005 to DY),and Shanghai Science and Technology Development Fund(Grant Nos.15DZ2291600 to MWW,16ZR1407100 to AD).We acknowledge the support of the NVIDIA Corporation for the donation of the Titan X GPU used in this study.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2019.04.003.
 Genomics,Proteomics & Bioinformatics2019年5期
Genomics,Proteomics & Bioinformatics2019年5期
- Genomics,Proteomics & Bioinformatics的其它文章
- Genomics,Proteomics&Bioinformatics
- MakeHub:Fully Automated Generation of UCSC Genome Browser Assembly Hubs
- VPOT:A Customizable Variant Prioritization Ordering Tool for Annotated Variants
- shinyChromosome:An R/Shiny Application for Interactive Creation of Non-circular Plots of Whole Genomes
- CircAST:Full-length Assembly and Quantification of Alternatively Spliced Isoformsin Circular RNAs
- CIRCexplorer3:A CLEAR Pipeline for Direct Comparison of Circular and Linear RNA Expression