
ILF2 cooperates with E2F1 to maintain mitochondrial homeostasis and promote small cell lung cancer progression
2019-02-16 06:07:04MengZhaoYahuiLiuJiaoChangJinQiRanLiuYongwangHouYanhuiWangXinweiZhangLuQiaoLiRen
Cancer Biology & Medicine 2019年4期
Meng Zhao, Yahui Liu, Jiao Chang, Jin Qi, Ran Liu, Yongwang Hou, Yanhui Wang, Xinwei Zhang,Lu Qiao, Li Ren
1Department of Clinical Laboratory; 2 Radiology Department, Tianjin Medical University Cancer Institute and Hospital,National Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, Tianjin, Tianjin’s Clinical Research Center for Cancer, National Human Genetic Resources Sharing Service Platform [2005DKA21300], Tianjin 300060,China; 3 Department of Immunology, Tianjin Key Laboratory of Cellular and Molecular Immunology, Key Laboratory of Educational Ministry of China, School of Basic Medical Sciences, Tianjin Medical University, Tianjin 300070, China
ABSTRACT Objective: Mitochondria play multifunctional roles in carcinogenesis. Deciphering uncertainties of molecular interactions within mitochondria will promote further understanding of cancer. Interleukin enhancer binding factor 2 (ILF2) is upregulated in several malignancies, however, much remains unknown regarding ILF2 in small cell lung cancer (SCLC). In the current study, we explored ILF2’s role in SCLC and demonstrated its importance in mitochondria quality control.Methods: Colony formation, cell proliferation, cell viability and xenograft studies were performed to examine ILF2’s role on SCLC progression. Glucose uptake, lactate production, cellular oxygen consumption rate and extracellular acidification rate were measured to examine the effect of ILF2 on glucose metabolism. RNA-sequencing was utilized to explore genes regulated by ILF2.E2F1 transcriptional activity was determined by dual luciferase reporter assay. Mitochondria quantification and mitochondrial membrane potential assays were performed to examine mitochondrial quality. Gene expression was determined by RT-qPCR,Western blot and IHC assay.Results: ILF2 promotes SCLC tumor growth in vitro and in vivo. ILF2 elevates oxidative phosphorylation expression and declines glucose intake and lactate production. Genome-wide analysis of ILF2 targets identified a cohort of genes regulated by E2F1. In consistent with this, we found ILF2 interacts with E2F1 in SCLC cells. Further studies demonstrated that suppression of E2F1 expression could reverse ILF2-induced tumor growth and enhanced mitochondria function. Significantly, expression of ILF2 is progressively increased during SCLC progression and high ILF2 expression is correlated with higher histologic grades, which indicates ILF2’s oncogenic role in SCLC.Conclusions: Our results demonstrate that ILF2 interacts with E2F1 to maintain mitochondria quality and confers SCLC cells growth advantage in tumorigenesis.
KEYWORDS E2F1; OXPHOS; mitochondria; metabolism; SCLC
Introduction
Tumors undergo metabolic reprogramming in order to meet energetic and biosynthetic demands1. However, there is no single tumor-specific metabolic phenotype because of the various and complex metabolic tumor landscapes that exist2.Tumor cells prefer to metabolize glucose through glycolysis;this observation led to the assumption that oxidative phosphorylation (OXPHOS) is suppressed in malignant states. However, recent studies report that cancers still demand OXPHOS, at least at low levels, to survive3.OXPHOS generates more ATP compared with glycolysis,which can be beneficial in environments with glucose and glutamine shortages.
Mitochondria are cytoplasmic rod-shaped organelles that are maternally inherited4. Importantly, they are hubs of bioenergy, biosynthesis and signaling transduction and the site where OXPHOS takes place4. Mitochondria control various cellular parameters including energy production,oxidation-reduction balance, ROS production, cytosolic calcium levels, contribution to biosynthetic precursors and initiation of apoptosis5. Multiple recent studies have highlighted the significance of mitochondria in all stages of malignant disease, including initiation, growth, survival and metastasis6. Furthermore, therapeutic targeting of one carbon metabolism, glutamine metabolism and complex I are currently being explored. However, these therapies are limited by varied responses in different tissue contexts and cancer types. Thus, it is of critical importance to fill in the gaps in our knowledge regarding mitochondrial biology.
Interleukin enhancer binding factor 2 (ILF2) is a transcription factor required for T-cell-specific expression of interleukin 2 (IL-2)7,8. It can also promote the formation of stable DNA-dependent protein kinase holoenzyme complexes on DNA9. Recent studies highlight its oncogenic role in a variety of malignant diseases, such as multiple myeloma, nonsmall cell lung cancer (NSCLC), pancreatic ductal adenocarcinoma and gastric cancer10-15. In the current study, we asked whether ILF2 influences SCLC progression and identified ILF2 as a novel regulator of OXPHOS and of mitochondrial homeostasis.
Materials and methods
Cell culture, patient samples and lentivirus infection
The human SCLC cell lines H446 and H82 were obtained from the Type Culture Collection of Chinese Academy of Sciences and maintained in culture according to their recommendations. Carcinoma and adjacent normal tissue samples were obtained from surgical specimens from SCLC patients. Samples were frozen in liquid nitrogen immediately after surgery. SCLC tissue assays were obtained from US Biomax, Inc. and subjected to immunohistochemical analysis according to standard protocols. Antibody was obtained from Santa cruz (ILF2) and Abcam (E2F1). All studies were approved by the Ethics Committee of the Tianjin Medical University Cancer Institute and Hospital, and informed consent was obtained from all patients.
ILF2-shRNA, E2F1-shRNA and control lentivirus were obtained from Shanghai Genepharma Co., Ltd. The ILF2-shRNA1 target sequence was 5′-CCACAGTTAAAG TTCTCATAA-3′. The ILF2-shRNA2 target sequence was 5′-GCTATCTTGCTTCTGAAATAT-3′. The E2F1-shRNA target sequence was 5′-CGCTATGAGACCTCACTGAAT -3′.Virus supernatant was incubated on target cells for 12 hours with 8 μg/ml polybrene, following the manufacturer’s instructions. Infected cells were selected in puromycin, as optimized for each cell line.
Colony formation assays
H446 or H82 cells were plated in triplicate in 6-well dishes using the appropriate growth media for each cell line. Media was replaced every two days. Colonies were counted after 7—10 days.
Cell proliferation assays
BrdU incorporation assays were performed as described previously16. Briefly, cells were seeded in triplicate in 96-well plates and incubated in complete medium for 24 h. Next,cell proliferation was determined using a Cell Proliferation ELISA, BrdU kit (Roche Applied Science, USA) according to the manufacturer’s instructions. Each point represents the mean value of three experiments including three replicates each.
For the MTS assays, pretreated SCLC cells were seeded in 96-well plates at a density of 5,000 cells/well. Twelve hours later, 10 μl of 0.5 mg/mL MTS reagent (Promega) was added to each well. Cells were incubated at 37°C for 2 h and then the Day 0 absorbance was detected at 570 nm on a μQuant Universal Microplate Spectrophotometer (Bio-Tek Instruments, Winooski, USA). Absorbance was detected at the same wavelength on Days 2, 3, 4 and 5. Absorbance values at each time point were normalized to the data from Day 0 in order to generate a proliferation curve.
Cell viability assays
SCLC cells were seeded in 96-well plates at a density of 5,000 cells/well. Twelve hours later, 10 μl of 0.5 mg/mL MTS reagent (Promega) was added to each well. Cells were incubated at 37°C for 2 h and then the absorbance was recorded.
Glucose and lactate determination
Glucose concentrations in cell culture media were determined using a Hitachi 7180 - Chemistry Analyzer. Cells were seeded in 6-well plates and media was collected at 0 h and 36 h. The glucose consumption rate was calculated as(Concentration0h— Concentration36h) × Volumemedia÷ cell number. Extracellular lactate was measured using the VITROS®5600 Integrated System. Cells were seeded in 6-well plates, and media was collected at 0 h and 36 h. The lactate production rate was calculated as (Concentration36h—Concentration0h) × Volumemedia÷ cell number.
RNA-sequencing and qRT-PCR
RNA isolation and qRT-PCR were performed as described previously16. Briefly, total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, USA). First-strand cDNA was synthesized from 2 μ g of total RNA using M-MLV reverse transcriptase (Invitrogen, Beijing, China). RNA-seq libraries were prepared using the Illumina RNA-seq Preparation Kit and sequenced on a HiSeq 2000 sequencer. Quantitative realtime PCR was performed using SYBR Green PCR Master Mix(Applied Biosystems). Reactions were performed on a 7500 Fast Real-Time PCR System (Applied Biosystems). The data are represented as 2-ΔΔCt values and are representative of at least three independent experiments. RT-PCR primer sequences are listed in Table 1.
Mitochondrial quantity
For mtDNA, total DNA was isolated using a Total DNA Extraction Kit (TIANGEN, Beijing, China). Relative levels of mtDNA copy number were determined by real-time PCR.The reactions were performed on a LightCycler®96 Real-Time PCR System (Roche). The data are displayed as 2-ΔΔCt values and are representative of at least three independent experiments. The following primers sets were used: Mt-Mito Forward: 5′-CACTTTCCACACAGACATCA-3′, Reverse:5′-TGGTTAGGCTGGTGTTAGGG-3′; B2M Forward: 5′-TG TTCCTGCTGGGTAGCTCT-3′, Reverse: 5′-CCTCCATGATGCTGCTTACA-3′. To determine mitochondrial mass, cells were stained with 100 nM MitoTracker Red (Invitrogen) at 37°C for 15 min and then analyzed by flow cytometry. The relative fluorescence intensity was record and represented as mitochondrial mass.

Table 1 qPCR primers
Mitochondrial membrane potential assays
Mitochondrial membrane potential was determined by incubating cells for 10 min at 37°C in medium containing 10 μg/mL of the mitochondrial membrane potential-sensitive fluorescent dye JC-1. Changes in the green/red fluorescence ratio were assessed by flow cytometry.
Cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR)measurement
OCR and ECAR were measured using an XF24 Extracellular Flux Analyzer (Seahorse Bioscience). In brief, pretreated SCLC cells were seeded in 24-well plates at a density of 1,000 cells/well and cultured overnight. Then, cells were washed with either OCR medium (containing 4.5 g/L glucose, 2 mM glutamine and 1 mM pyruvate) or ECAR medium (containing 2 mM glutamine and no pyruvate or glucose) and incubated in a CO2-free incubator at 37°C for 1 h to allow for temperature and pH equilibration prior to loading into the XF24 apparatus. XF assays consisted of 3 cycles of: Mix (3 min), Wait (2 min), and Measure (3 min),including 3 basal rate measurements prior to the first injection and 3 rate measurements after each injection. ECAR was measured under baseline conditions and after treatment with glucose (100 mM), oligomycin (100 μM) and 2-deoxy glucose (2-DG; 500 mM). OCR was measured under baseline conditions and after treatment with Oligomycin (100 μM),FCCP (100 μM) and Rotenone/Antimycin (50 μM). Values were normalized to 1 × 104cell counts. Values are presented as the mean ± standard error.
Immunoprecipitation and Western blot
H446 cellular extracts were obtained by pelleting cells at 13,000 rpm for 10 min at 4°C and then incubating cells in lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5%NP40) for 30 min at 4°C. For immunoprecipitation, extracts were incubated with the indicated antibody overnight at 4°C.Next, protein A agarose beads were added, and the mixture was incubated for 3 h. Beads were washed five times, and then, precipitated proteins were resolved by 10% SDS-PAGE and transferred to acetate cellulose membranes. Western blotting was performed as previously described17. Briefly,antibody binding was revealed using an HRP-conjugated anti-rabbit IgG or anti-mouse IgG secondary antibody(Sigma). Antibody complexes were detected using the Immobilon Western Chemiluminescent HRP Substrate(Millipore) and the Tanon 6200 Luminescent Imaging Workstation (Tanon Science & Technology Co., Ltd.,Shanghai, China). Antibodies were obtained from Cell Signaling (E2F1), Santa Cruz (ILF2) and Sigma (β-actin).
Proximity ligation assay (PLA)
PLA was performed using a Duolink®In Situ Detection Reagents Red kit (Sigma). In brief, cells were fixed and permeabilized, then incubated with antibody ILF2 (1 : 1,000)and E2F1 (1 : 1,000). After incubated with secondary antibody,ligase buffer and DNA amplification buffer were added and incubated sequentially. Then nucleus was stained with DAPI.
Immunohistochemistry
Murine tumors were fixed in 10% neutral buffered formalin for 24 h. Then, tumors were processed for paraffin embedding. Of note, 5 mmol/L sections were used for hematoxylin and eosin staining and immunohistochemistry.Unstained sections were deparaffinized, rehydrated and stained for Ki67 (Abcam), cleaved caspase 3 (Cell Signaling).
Dual luciferase reporter assay
The E2F1 transcriptional activity reporter plasmid was obtained from the Cignal Reporter Assay Kit (CCS-003L,QIAGEN). Luciferase activity was measured using a dual luciferase kit (Promega, Madison, WI) according to the manufacturer’s protocol. Each experiment was performed in triplicate and was repeated at least three times.
SCLC xenograft models
Female athymic nude mice were purchased from the Academy of Military Medical Science (Beijing, China). All mouse studies were approved by the Animal Ethics Committee of Tianjin Medical University. All animals were 4 — 6 weeks of age at the time of injection. H446-scramble or H446-shILF2-1 cells were trypsinized, washed, resuspended in Hank’s Balanced Salt Solution (HBSS; Gibco) and subcutaneously injected into the right flank of each mouse(5 × 106cells/animal).
Statistical analysis
Data are reported as the mean ± SD. Biochemical experiments were performed in triplicate, and a minimum of three independent experiments were evaluated. Differences were assessed for statistical significance using an unpaired two-tailed t-test. Significant p values are denoted as follows:*P < 0.05; ** P < 0.01.
Results
ILF2 promotes SCLC cell proliferation and tumorigenesis
To explore the functional role of ILF2 in SCLC progression,we first suppressed its expression in two SCLC cell lines(H446 and H82) using shRNAs. Knockdown efficiency was confirmed by RT-qPCR and Western blotting (Figure 1A and 1B). Cell morphology and viability remained unchanged upon ILF2 inhibition (Supplementary Figure S1A and S1B).Interestingly, colony formation assays revealed that shILF2 cells produced fewer colonies compared with scrambled control shRNA-treated cells (Figure 1C and 1D).Furthermore, ILF2 knockdown resulted in decreased cell proliferation rates in both H446 and H82 cells(Supplementary Figure S1C and S1D). BrdU is an analog of the DNA precursor thymidine. Incorporation of BrdU reflects the proliferative ability of cells. Our results demonstrated that ILF2 knockdown suppressed BrdU incorporation in H446 and H82 cells (Figure 1E). These results indicated that ILF2 may enhance proliferation in SCLC cells. To further confirm this hypothesis, we overexpressed ILF2 in H446 and H82 cells (Figure 1F and 1G). As expected, ILF2 upregulation resulted in enhanced colony formation (Figure 1H and 1I). Cell proliferation and BrdU incorporation rates were also increased after ILF2 overexpression (Supplementary Figure S1E and S1F, Figure 1N). We also established a xenograft model to examine the impact of ILF2 on tumorigenesis in vivo. The results showed that ILF2 knockdown resulted in reduced tumor volume and weight in vivo (Figure 1K, 1L and 1M). In addition, we examined Ki67 and cleaved caspase 3 expression in ILF2 knockdown and scrambled shRNA control tumors. We found that ILF2 knockdown inhibited Ki67 expression(Figure 1N) but had no impact on levels of cleaved caspase 3(Supplementary Figure S1G). This finding is consistent with our data showing that ILF2 promotes cell proliferation.Taken together, our results demonstrate that ILF2 plays a positive role in SCLC cell proliferation and tumorigenesis.

Figure 1 ILF2 promotes SCLC cell proliferation and tumorigenesis. (A and B) Two short hairpin RNAs were used to knock down ILF2 expression in H446 and H82 cells. RT-qPCR and Western blot were used to confirm the knockdown efficiency. (C and D) H446 and H82 cells infected with shILF2 or scrambled shRNA were assessed by colony formation assay. Representative graphs showing the colony formation capacity of H446 cells are shown in panel (C). (E) H446 and H82 cells infected with shILF2-1, shILF2-2 or scrambled shRNA were analyzed by BrdU incorporation assays. (F and G) RT-qPCR and Western blot were used to assess ILF2 expression levels in H446 and H82 cells infected with either an ILF2-overexpressing or a vector control lentivirus. (H and I) H446 and H82 cells infected with ILF2-overexpressing or vector control lentivirus were assessed by colony formation assay. Representative graphs showing the colony formation capacity of H446 cells are shown in panel (H). (J) H446 and H82 cells infected with ILF2-overexpressing or control vector lentivirus were analyzed by BrdU incorporation assays. (K, L and M) Female nude mice were injected with H446-scramble or H446-shILF2-1 cells (5 × 106 cells/animal). Three weeks after injection, the mice were euthanized. Tumor burden was evaluated based on tumor volumes and weights (n = 8). (N) Xenograft tumor sections were stained with H&E or an anti-Ki67 antibody. *, P < 0.05, **, P < 0.01.
ILF2 enhances OXPHOS and suppresses aerobic glycolysis in SCLC cells
Cancer cells reprogram their metabolic phenotype in order to adapt to a heterogeneous microenvironment2. We were interested in whether ILF2 could affect cell metabolism as well. Glucose is a major carbon and energy source for tumor growth. We thus examined the impact of ILF2 on glucose utility. Our results demonstrated that ILF2 downregulation resulted in increased glucose uptake in H446 and H82 cells(Figure 2A). The end product of glucose metabolism can either be lactate or, upon full oxidation, CO2 and H2O. We found that ILF2 inhibition induced greater lactate production in H446 and H82 cells (Figure 2B), indicating enhanced aerobic glycolysis. To confirm this hypothesis, we examined the extracellular acidification rate (ECAR) in H446 and H82 cells infected with shILF2-1, shILF2-2 or scrambleshRNA. Our results demonstrated that ILF2 knockdown significantly enhanced glycolysis and reduced glycolytic reserves in H446 and H82 cells (Figure 2C, 2E and 2G). We then examined OXPHOS by measuring the cellular oxygen consumption rate (OCR). Our results demonstrated that ILF2 knockdown significantly suppressed both basal and maximal respiration in H446 and H82 cells (Figure 2D, 2F and 2H ). Consistent with the above data, ILF2 overexpression resulted in suppressed glucose uptake and lactate production in H446 and H82 cells (Figure 2I and 2J).Reduced glycolysis and increased glycolytic reserves were both observed following ILF2 overexpression (Figure 2K, 2M and 2O). Enhanced basal and maximal respiration were also observed (Figure 2L, 2N and 2P). Taken together, these data indicate that ILF2 participates in glucose metabolism and confers SCLC cells with enhanced OXPHOS capacity.
ILF2 interacts with E2F1 in vivo and regulates its transcriptional activity
To further explore the molecular function of ILF2, we performed transcriptomic sequencing in shILF2-1 cells and scrambled shRNA control cells. ILF2 knockdown resulted in the downregulation of 75 genes and upregulation of 74 genes(Figure 3A and 3B). We then performed Gene Ontology(GO) analysis, KEGG pathway analysis and Gene Set Enrichment Analysis (GSEA) (Supplementary Figure S2A and S2B ) on these differentially expressed genes.Surprisingly, the E2F target gene set was found to be enriched in ILF2 knockdown cells (Figure 3C). These results indicate that ILF2 may participate in the regulation of E2F1 activity.We first examined whether alterations in ILF2 expression or activity could affect E2F1 expression. The results demonstrated that neither inhibition nor overexpression of ILF2 altered E2F1 expression (Figure 3D and 3E). As ILF2, in addition to E2F1, has transcriptional activity, we proposed that ILF2 may interact with E2F1 to direct downstream target expression. To test this hypothesis, we overexpressed FLAGE2F1 in H446 cells and performed coimmunoprecipitation using a FLAG antibody followed by Western blotting using an anti-ILF2 antibody. The results demonstrated that ILF2 is efficiently coimmunoprecipitated with E2F1 (Figure 3F). To further validate the results, we performed a proximal ligation assay (PLA) to detect protein interaction in situ. In consistent with Co-IP assay, PLA demonstrate ILF2 interacted with E2F1 in cell nucleus (Figure 3G). Next, we examined whether ILF2 could regulate the transcriptional activity of E2F1. We used a dual-luciferase reporter system and found that ILF2 knockdown significantly suppressed the transcriptional activity of E2F1 while ILF2 overexpression enhanced its activity (Figure 3H and 3I). Taken together, these results suggest that ILF2 interacts with E2F1 and regulates E2F1 transcriptional activity.
ILF2 maintains mitochondrial quantity and membrane potential in SCLC

Figure 2 ILF2 enhances OXPHOS and suppresses aerobic glycolysis in SCLC cells. (A and B) H446 and H82 cells were infected with shILF2 or scrambled shRNAs. Then, glucose uptake and lactate production were measured over the subsequent 36 h. (C-F) Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) were assessed in H446 and H82 cells infected with shILF2 or scrambled shRNAs. (G and H) Glycolysis, glycolytic capacity, glycolytic reserve, basal respiration and maximal respiration in H446 and H82 cells were compared using data from Panels C-F. (I and J) H446 and H82 cells were infected with ILF2-overexpressing or vector control lentivirus. Then,glucose uptake and lactate production were measured over the next 36 h. (K-P) Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) were assessed in H446 and H82 cells infected with ILF2-overexpressing or vector control lentivirus. (O and P)Glycolysis, glycolytic capacity, glycolytic reserve, basal respiration and maximal respiration of H446 and H82 cells were compared using data from Panels K-N. *, P < 0.05, * *, P < 0.01.
Recent studies highlight the role of E2F1 in mitochondrial quality control and metabolism18,19. The above data indicate that ILF2 interacts with E2F1. We wanted to investigate whether ILF2 also participates in mitochondrial function. We first investigated whether ILF2 could affect mitochondrial quantity. We observed decreased mtDNA in ILF2 knockdown cells compared with scrambled shRNA control cells (Figure 4A). Furthermore, ILF2 knockdown also resulted in decreased mitochondrial mass in H466 and H82 cells (Figure 4B). These results indicate that ILF2 participates in mitochondrial homeostasis. Inner mitochondrial membrane potential (ΔΨm) is an indicator of mitochondrial energy production and homeostasis. We thus examined the impact of ILF2 loss on ΔΨm using the JC-1 dye. Our results showed that ILF2 knockdown significantly reduced ΔΨm in both H446 and H82 cells (Figure 4C). These data indicate that ILF2 knockdown cells exhibit low-quality mitochondria.
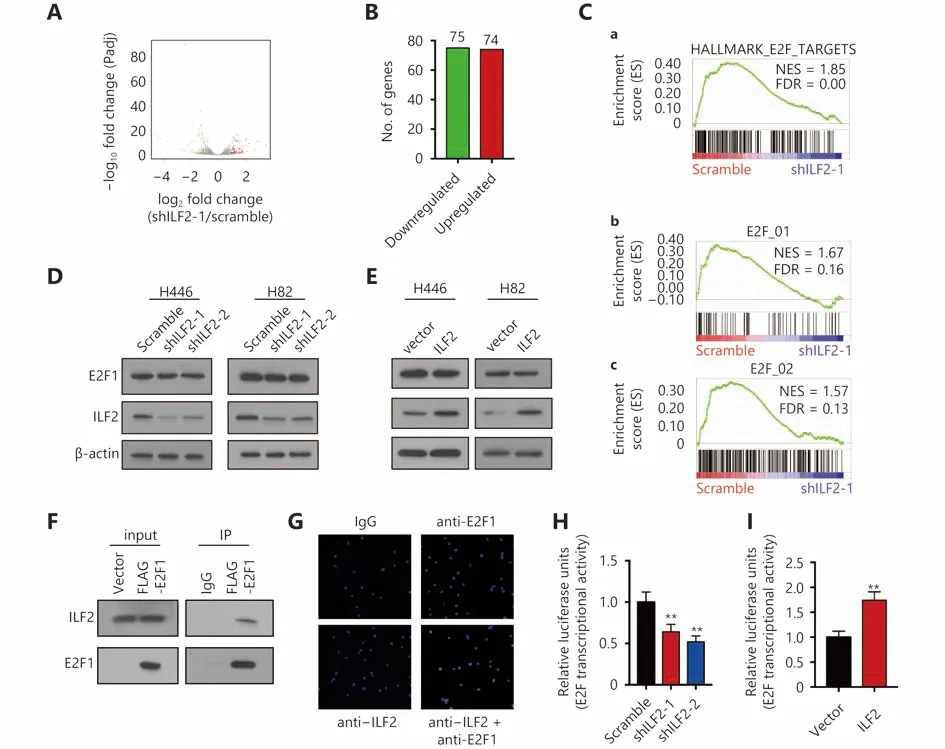
Figure 3 ILF2 interacts with E2F1 in vivo and regulates its transcriptional activity. (A) Volcano plot showing the transcriptomic expression profiles of shILF2-1 and scrambled shRNA-treated H446 cells. Red and green dots represent genes significantly upregulated and downregulated in shILF2-1 cells (P < 0.05), respectively. The gray dots represent insignificantly changed genes with P > 0.05. (B) Number of upregulated and downregulated genes in shILF2-1 cells. (C) GSEA analysis of gene sets enriched in shILF2-1 cells. (D and E) E2F1 expression was analyzed in ILF2-knockdown and ILF2-overexpressing H466 and H82 cells via Western blot. (F) Coimmunoprecipitation assay in H446 cells using an anti-FLAG followed by immunoblotting (IB) with antibodies against the indicated proteins. (G) PLA assay was performed using ILF2 and E2F1 antibodies in H446 cells. (H and I) Dual-luciferase reporter assay for E2F1 transcriptional activity. *, P < 0.05, * *, P < 0.01.
Then, we examined whether ILF2 inhibition could also alter electron transport chain (ETC) genes, such as NDUFS6,UQCRB, COX7B and ATP5G1. In agreement with our hypothesis, all of these genes were downregulated in ILF2 knockdown cells (Figure 4D and 4E). The major function of the ETC is to effectively produce ATP in order to meet cellular energy demands. We then examined whether ILF2 inhibition could affect ATP quantity in SCLC cells. Our results demonstrated that total ATP quantities remained unchanged after ILF2 knockdown (Figure 4F). Cells produce ATP via two pathways: OXPHOS and glycolysis. We hypothesized that cells may compensate for decreased ETC function by enhancing glycolysis. To address this hypothesis,we incubated cells with 2-Deoxy-D-glucose (2-DG), a glucose analog that inhibits glycolysis, in order to see whether 2-DG could alter total ATP levels. In keeping with our hypothesis, 2-DG-treated, ILF2-inhibited cells displayed decreased levels of ATP (Figure 4F).
Furthermore, ILF2 overexpression resulted in increased mtDNA and mitochondrial mass in H446 and H82 cells(Figure 4G and 4H). Increased ΔΨm and upregulation of ETC genes were also observed in cells overexpressing ILF2(Figure 4I, 4J and 4K). Taken together, these data demonstrate that ILF2 plays a role in maintaining mitochondrial homeostasis in SCLC cells.
ILF2-induced OXPHOS and enhanced proliferation is E2F1 dependent

Figure 4 ILF2 maintains SCLC mitochondrial quantity and membrane potential. (A) The relative quantity of mtDNA in H446 and H82 cells infected with shILF2 or scrambled shRNAs was determined by real-time qPCR. (B) Mitochondrial mass in H446 and H82 cells infected with shILF2 or scrambled shRNAs. (C) JC-1 green fluorescence intensity in H446 and H82 cells infected with shILF2 or scrambled shRNAs. (D and E) ETC gene expression levels were evaluated by RT-qPCR in H446 and H82 cells infected with shILF2 or scrambled shRNAs. (F) H446 and H82 cells infected with shILF2 or scrambled shRNAs were treated with different concentrations of 2-DG for 24 h and then total ATP was measured. (G) Relative quantity of mtDNA in H446 and H82 cells infected with ILF2-overexpressing or vector control lentivirus. (H)Mitochondrial mass in H446 and H82 cells infected with ILF2-overexpressing or vector control lentivirus. (I) JC-1 green fluorescence intensity in H446 and H82 cells infected with ILF2-overexpressing or vector control lentivirus. (J and K) ETC gene expression levels were evaluated by RT-qPCR in H446 and H82 cells infected with ILF2-overexpressing or vector control lentivirus. *, P < 0.05, * *, P < 0.01.
We sought to test whether the functional role of ILF2 in cell proliferation and glucose metabolism is E2F1-dependent. We first knocked down E2F1 expression in ILF2-overexpressing H446 and H82 cells (Figure 5A). Next, we performed colony formation and BrdU incorporation assays. Our results demonstrated that E2F1 knockdown significantly decreased colony numbers and BrdU incorporation rates (Figure 5B and 5C ). Furthermore, E2F1 downregulation enhanced glucose uptake and lactate production in H446 and H82 cells infected with ILF2-expressing lentivirus (Figure 5D and 5E).Suppression of E2F1 also reversed ILF2-induced mitochondrial enhancements (Figure 5F and 5G). In addition, E2F1 knockdown promoted aerobic glycolysis and suppressed OXPHOS in H446 and H82 cells infected with an ILF2-overexpressing lentivirus (Figure 5H-5M). Taken together,these results indicate that the role of ILF2 in tumorigenesis and glucose metabolism is dependent on E2F1 expression.
ILF2 is upregulated in SCLC and correlates with pathological grade

Figure 5 ILF2-induced enhancements in OXPHOS and proliferation are E2F1 dependent. (A) ILF2-overexpressing H446 cells were infected with short hairpin RNAs targeting E2F1. Knockdown efficiency was determined by RT-qPCR. H446-ILF2 and H82-ILF2 cells infected with shE2F1 and scramble control RNAs were assessed by colony formation (B) and BrdU incorporation (C) assays . Glucose uptake rate (D) and lactate production (E) were measured in H446-ILF2 and H82-ILF2 cells infected with shE2F1 and scramble control RNAs. (F) Relative quantity of mtDNA in H446 and H82 cells infected with ILF2-overexpressing or vector control lentivirus. (G) Mitochondrial mass in H446 and H82 cells infected with ILF2-overexpressing or vector control lentivirus. (H-M) Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) in H446-ILF2 and H82-ILF2 cells infected with shE2F1 and scramble control RNAs. *, P < 0.05, * *, P < 0.01.
To further characterize the role ILF2 in SCLC progression,we obtained 15 SCLC samples with paired adjacent normal lung tissues. We also collected 5 normal lung samples and 5 lung tissue samples with benign disease. ILF2 mRNA expression levels were analyzed in all samples by RT-qPCR.We found that ILF2 is upregulated in malignant samples compared with normal and benign tissues (Figure 6A).Additionally, ILF2 is upregulated in SCLC samples compared with their paired samples. We then we examined ILF2 protein expression via immunohistochemical staining of a tissue array consisting of SCLC samples of various grades.Remarkably, we found that ILF2 is significantly upregulated in SCLC compared with normal tissue, adjacent tissue,tuberculosis samples, and inflammatory pseudotumor samples (Figure 6C). We also demonstrate that ILF2 is upregulated in high SCLC pathological grades, which indicates that ILF2 expression progressively increases during SCLC progression (Figure 6C and 6D). Although we did not produce data demonstrating a positive correlation between ILF2 expression and SCLC prognosis, public data from online databases revealed that high ILF2 expression levels correlated with poor NSCLC prognosis (Supplementary Figure S3). These data are consistent with the oncogenic role of ILF2 in SCLC.
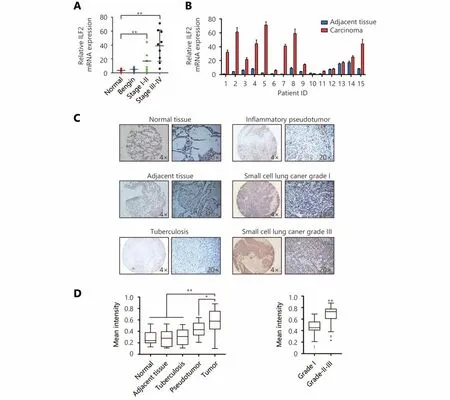
Figure 6 ILF2 is upregulated in SCLC and correlates with pathological grade. (A) ILF2 levels were analyzed by qRT-PCR in cDNA from 15 SCLC patient samples representing all disease stages and grades, 5 lung tissues with benign disease and 5 normal lung samples. (B) ILF2 levels were analyzed by qRT-PCR in 15 SCLC samples with paired adjacent normal lung tissues. (C and D) Immunohistochemical staining of tissue arrays. The mean staining intensity was calculated using Image-Pro Plus software. Representative IHC images showing ILF2 and E2F1 expression in SCLC tissue microarray (C). *, P < 0.05, * *, P < 0.01.
Discussion
In this study, we demonstrated that ILF2 plays an oncogenic role in SCLC. ILF2 expression is upregulated in SCLC and promotes SCLC cell proliferation in vitro and growth in vivo.Furthermore, in addition to its role in DNA damage response, we found that ILF2 also regulates mitochondrial function, especially OXPHOS. In combination with transcriptomic analysis and Co-IP, we demonstrated that ILF2 interacts with E2F1, a transcriptional activator, which has recently been reported to regulate mitochondrial function. We found that the ILF2-E2F1 interaction is crucial for the ILF2-induced enhancement of mitochondrial OXPHOS.
SCLC is a clinically and histologically distinct type of lung cancers. Because of its poor prognosis, SCLC remains a frustrating disease to research and to treat20. Thus, it is important to identify new SCLC driver genes and therapeutic targets. ILF2 was first identified as a transcription factor required for T-cell-specific expression of the interleukin 2 gene and is now known to function in a heterodimeric complex with ILF37,8. Further studies have demonstrated its role in mRNA and microRNA processing21,22. ILF2 also participates in cell growth and tumor progression14.However, its role in SCLC is largely unknown. We observed that ILF2 is upregulated in SCLC compared with normal and adjacent tissues and that the expression of ILF2 correlates with SCLC pathological grade. In addition, ILF2 promotes SCLC cell proliferation in vitro and tumor growth in vivo.These results indicate that ILF2 plays an oncogenic role in SCLC progression.
We expanded our analysis of the function of ILF2 to the field of metabolism. We found that ILF2 directs the full oxidation of glucose to H2O and CO2via mitochondrial respiration, rather than promoting glycolysis. This phenomenon confers a growth advantage to SCLC cells under glucose shortage conditions. We speculated that ILF2 exerted its metabolic function through its interaction with E2F1. However, whether ILF2 also utilizes its mRNA processing functionality to affect metabolic gene expression remains to be answered.
E2F gene family members are bona fide transcription factors that are involved in controlling the transition from G1 to S phase23. Notably, E2F is upregulated in a variety of malignant diseases. However, E2F binds thousands of genes,and thus, other biologic processes may be overlooked by researchers. Recent studies indicate that E2F regulates the expression of mitochondria-associated genes24. E2F regulates mitochondrial function in Drosophila and is regulated by NRF1 and NRF2, which are key factors involved in mitochondrial biogenesis25. Our observation that E2F1 loss suppresses mitochondrial function is consistent with these findings. Furthermore, we found that ILF2 interacts with E2F1 in vivo. Loss of ILF2 suppresses E2F1 transcriptional activity and overexpression of ILF2 reverses these effects.RNA-sequencing data showed that ILF2 target genes significantly overlap with E2F1 target genes. We proposed that ILF2 cooperates with E2F1 to regulate mitochondrial function.
The discovery of high rates of aerobic glycolysis in cancer cells by Otto Warburg led to the intensive study of cancer metabolism. Now, we know that glucose metabolism encompasses not only glycolysis but also other pathways such as the pentose phosphate pathway (PPP), the hexosamine pathway, glycogenesis and the serine biosynthesis pathway26.In addition, cancer cells do not always prefer high rates of glycolysis. Some cancer types prefer OXPHOS and other cancer types may prefer different metabolic phenotypes at different stages of progression27-32. Little is known about the preferred metabolic phenotype in SCLC. In the current study, we showed that OXPHOS confers a growth advantage to SCLC cells both in vitro and in vivo. We also showed that ILF2 is upregulated during the progression of SCLC, which indicates that SCLC prefers an OXPHOS status. However,further investigation is required in order to confirm this hypothesis.
Conclusions
In summary, our results reveal that ILF2 plays an oncogenic role in SCLC progression. Importantly, ILF2 interacts with E2F1 to maintain mitochondrial quality and function, which confer a growth advantage to SCLC cells. Moreover, our experiments highlight the significance of OXPHOS in SCLC.However, the precise molecular mechanism by which ILF2 regulates metabolism has yet to be elucidated.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 81602026) and the Natural Science Foundation of Tianjin (Grant No. 18JCQNJC81600 and 18JCZDJC32600).
Conflict of interest statement
No potential conflicts of interest are disclosed.
 Cancer Biology & Medicine2019年4期
Cancer Biology & Medicine2019年4期
- Cancer Biology & Medicine的其它文章
- Interpretation of breast cancer screening guideline for Chinese women
- Breast cancer screening guideline for Chinese women
- Erratum to Simultaneous inhibition of PI3Kα and CDK4/6 synergistically suppresses KRAS-mutated non-small cell lung cancer
- The correlation and overlaps between PD-L1 expression and classical genomic aberrations in Chinese lung adenocarcinoma patients: a single center case series
- Nomogram based on albumin-bilirubin grade to predict outcome of the patients with hepatitis C virus-related hepatocellular carcinoma after microwave ablation
- Omics-based integrated analysis identified ATRX as a biomarker associated with glioma diagnosis and prognosis