
Tobacco, air pollution, environmental carcinogenesis, and thoughts on conquering strategies of lung cancer
2019-02-16 06:07:00GuangbiaoZhou
Cancer Biology & Medicine 2019年4期
Guangbiao Zhou
State Key Laboratory of Molecular Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, China
ABSTRACT Each year there will be an estimated 2.1 million new lung cancer cases and 1.8 million lung cancer deaths worldwide. Tobacco smoke is the No.1 risk factors of lung cancer, accounting for > 85% lung cancer deaths. Air pollution, or haze, comprises ambient air pollution and household air pollution, which are reported to cause 252,000 and 304,000 lung cancer deaths each year,respectively. Tobacco smoke and haze (hereafter, smohaze) contain fine particles originated from insufficient combustion of biomass or coal, have quite similar carcinogens, and cause similar diseases. Smohaze exert hazardous effects on exposed populations, including induction of a large amount of mutations in the genome, alternative splicing of mRNAs, abnormalities in epigenomics, initiation of tumor-promoting chronic inflammation, and facilitating immune escape of transformed cells. Tackling smohaze and development of multi-targets-based preventive and therapeutic approaches targeting smohaze-induced carcinogenesis are the key to conquer lung cancer in the future.
KEYWORDS Lung cancer; tobacco smoke; air pollution; smohaze; carcinogenesis
Introduction
Lung cancer is the most common cause of cancer death worldwide, with 2.1 million new cases and 1.8 million deaths predicted in 20181. Lung cancer is also No. 1 cancer killer in China2(Figure 1). Lung cancer is categorized by cell type into small-cell lung cancer (SCLC; accounting for 15% of all lung cancer cases) and non-small-cell lung cancer (NSCLC;85%), whereas NSCLC can be divided into lung adenocarcinoma (LUAD; 40%), lung squamous cell carcinoma (LUSC; 30%), and large cell lung cancer (LCLC;15%). The past two decades have witnessed enormous progress in development of targeted and immune therapies,which significantly improve the clinical outcome of the patients3. Targeted therapies include inhibitors of tyrosine kinases EGFR4, MET5, HER26, and BRAF V6007, inhibitors of fusion proteins involved ALK8, ROS19, RET10,11, and TRK12,antibodies against VEGF13, and others. Immune therapies include antibodies against cytotoxic T-cell lymphocyte-4(CTLA-4)14, programmed cell death 1 (PD-1) receptor15, and PD-1 ligand 1 (PD-L1)16.
However, drug resistance will eventually occur in patients treated with these therapies17, leading to treatment failure of the patients. Currently, the 5-year overall survival of lung cancer of all stage combined is only 18.6%18and 7%19,respectively. Therefore, new strategies remain urgent needs to conquer lung cancer. It is well-known that a large majority of patients suffering from lung cancer have their roots in unhealthy life style and exposure to environmental factors,hence much more attentions should be paid to the causes of lung cancer and related lung carcinogenesis when considering curative approaches for this deadly disease.
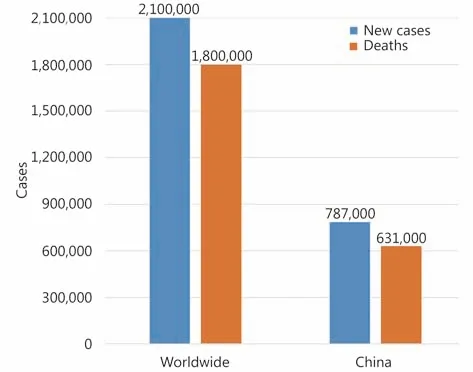
Figure 1 Estimated new lung cancer cases and lung cancer deaths worldwide1 and in China2 in 2018.
Tobacco smoke: the No. 1 risk factor of lung cancer
Tobacco smoke is the single biggest public health threat the world is currently facing. It is estimated that there are 1.1 billion smokers worldwide, and around 8 million people are killed by tobacco smoke each year globally. Among these deaths, more than 7 million are the result of direct tobacco use while around 1.2 million are the result of non-smokers being exposed to second-hand smoke20-22. Tobacco use is responsible for approximately 22% of cancer deaths worldwide23. Recent studies show that approximately 15% to 24% of lifetime smokers will get lung cancer24, and tobacco smoking accounts for more than 85% of lung cancer worldwide25.
During smoking, tobacco is heated to 880°C or higher, and more than 8,000 compounds have been identified in tobacco and tobacco smoke. More than 70 carcinogens and more than 20 lung carcinogens have been identified, which fall into group 1 (carcinogenic to humans), group 2A (probably carcinogenic to humans), and group 2B (possibly carcinogenic to humans) of carcinogen classifications of the International Agency for Research on Cancer (IARC) of the World Health Organization (WHO). Group 1 carcinogens found in tobacco smoke include polycyclic aromatic hydrocarbons [PAHs; e.g., benzo (a) pyrene, BaP], Nnitrosamines [e.g., N’-nitrosonornicotine; 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, NNK],aromatic amines (e.g., 2-naphthylamine, 4-aminobiphenyl),heterocyclic aromatic amines [e.g., 2-amino-3-methylimidazo (4,5-f) quinoline], aldehydes (e.g.,formaldehyde), volatile hydrocarbons (e.g., benzene),miscellaneous organic compounds (e.g., vinyl chloride,ethylene oxide), heavy metals and inorganic compounds[e.g., arsenic, beryllium, nickel, chromium (hexavalent),cadmium, radioisotope polonium-210], and others24,26.
Cigarette smoking was linked to lung cancer in 1964 by the Surgeon General of U.S. Department of Health and Human Service in the first Surgeon General’s Report. This linking has had an enormous positive effect on public health, in that in U.S. male smoking prevalence has decreased from 51.1% to the current 21.6%, whereas prevalence in females diminished from 33.3% to 16.5%24. In China, male and female smoking prevalence has also began to decrease27. Due to reduced tobacco use, lung cancer death rates in U.S. declined 45%from 1990 to 2015 among males and 19% from 2002 to 2015 among females1. However, there will be an estimated 228,150 new lung cancer cases and 142,670 lung cancer deaths in 2019 in U.S.28, and there will be an estimated 2,540,842 new lung cancer cases and 2,145,215 lung cancer deaths worldwide in 202529. Of note, no therapeutics so far are designed to target tobacco smoke-induced lung carcinogenesis, which remains to be elucidated.
Air pollution represents the second risk factor of lung cancer
Clean air is a basic requirement of life. Air pollution, or haze,is a diverse mixture of pollutants that originated from anthropogenic and natural sources, is comprised of particulate matters (PM), gases (e.g., sulfur oxides, carbon monoxide, ozone), organic compounds (e.g., PAHs; resin acids; anhydrous sugars; lignin pyrolysis products; hopanes),metals (e.g., lead, vanadium, and nickel), microbes, and others30-32. The main PM in the environment are PM smaller than 10 μm in diameter (PM10) and PM smaller than 2.5 μm in diameter (PM2.5), and air pollution can be divided into ambient air pollution (AAP) and household air pollution(HAP), and is a global environmental health risk that affects the populations in developed and developing countries alike,and 91% of the world population is living in places where the WHO air quality guidelines levels (i.e. annual mean values of 20 μg/m3for PM10and 10 μg /m3for PM2.5)33are not met34.WHO also sets guidelines for the protection of public health from risks due to a number of chemicals commonly present in indoor air {i.e. benzene, carbon monoxide, formaldehyde,naphthalene, nitrogen dioxide, polycyclic aromatic hydrocarbons [especially benzo (a) pyrene], radon,trichloroethylene and tetrachloroethylene}35. AAP in both cities and rural areas is estimated to cause 4.2 million premature deaths worldwide each year34, and 3.8 million people a year die prematurely from illness attributable to the HAP caused by the inefficient combustion of solid fuels and kerosene for cooking36. It is estimated that there are 304,00037and 252,00034lung cancer deaths that are attributed to HAP and AAP each year worldwide,respectively.
Xuanwei (XW) City in Yunnan Province of China provides an example of the epidemiological association between PM10,PM2.5and lung cancer38-40. This city has a large deposit of smoky coal and until the 1970s, local residents used smoky coal in unvented indoor firepits for domestic cooking and heating, all processes that release high concentrations of PM10and PM2.5. These airborne particles contain high concentrations of PAHs including BaP and polar compounds that are highly mutagenic40. Lung cancer incidence in XW is among the highest in China38,40, and a reduction in lung cancer morbidity was noted in the 1990s after stove improvement in central XW, supporting the association between air pollution and lung cancer41. These findings had been cited in the IARC monograph classifying HAP as“carcinogenic to humans (group 1)”42. On the other hand,Raaschou-Nielsen et al.43showed that the risk of lung cancer rises by 18% for every increase of 5 μg/m3of PM2.5in the environment, and the risk increases by 22% for every increase of 10 μg/m3in PM10. AAP has also been classified as group 1 carcinogen in humans by IARC43. LUAD, LUSC, SCLC, and bronchiolo-alveolar carcinoma (BAC) represent the most frequently seen lung cancer subtypes in air pollution regions38,44.
Tobacco smoke and haze (smohaze)are responsible for more than 90% of lung cancer
Tobacco smoke and haze bear many common characteristics in causing lung cancer (Table 1). For example, the fine particles in the two factors are PM2.5, which are mainly originated from insufficient combustion of biomass or coal.Though they may contain different chemical components,tobacco smoke and haze have quite similar main carcinogens,e.g., PAHs, heterocyclic compounds, N-nitrosamines,aromatic amines, heterocyclic aromatic amines, aldehydes,phenolic compounds, volatile hydrocarbons,nitrohydrocarbons, miscellaneous organic compounds,metals, and others (Table 1). PM2.5are inhaled, and cause diseases such as respiratory infections, cardiovascular disease,chronic obstructive pulmonary disease (COPD), and cancers20,21,34,37. Tobacco smoke and haze can cause all types of lung cancer (Table 1). Furthermore, these two factors are responsible more than 90% of lung cancer. Therefore, to elucidate the environmental lung carcinogenesis and to develop effective chemopreventive and therapeutic approaches to conquer lung cancer, tobacco smoke and haze should be treated as a single risk factor, smohaze (for tobacco smoke and haze).
Why smohaze
Treating the two risk factors as one, smohaze, may result in some positive effects in prevention and treatment of related disease. First, the health hazards of tobacco smoke and haze are currently separately assessed, which may lead to inconsistent results. For example, it is estimated that > 85%(1,530,000) of the 1.8 million lung cancer deaths areattributed to tobacco smoke, and other reports show that 252,000 and 304,000 patients may die from AAP- and HAPcaused lung cancer, respectively. Obviously, deaths from these factors exceed the total lung cancer deaths worldwide.Under the concept of smohaze, the more accurate data will be obtained from the related agencies. Second, when considering preventive medicine, the two factors should be tackled simultaneously. Combination of the two would be helpful for the public health domain to develop more effective strategies to tackle the public health problems, in that anti-tobacco campaign and anti-haze efforts could be propagated simultaneously. Third, the emerging concept of smohaze may create additional opportunities for the elucidation of environmental lung carcinogenesis and development of effective preventive and therapeutic approaches to conquer lung cancer.

Table 1 Comparison of tobacco smoke and haze
Smohaze-induced lung carcinogenesis
Efforts have been made to dissect tobacco smoke-induced tumorigenesis and air pollution in promoting lung cancer.These works show that smohaze exerts comprehensive effects on humans to trigger and promote lung cancer (Figure 2).
Genomic mutations
Carcinogens and nucleotide substitutions
As compared with counterpart normal controls, cancer genomes usually have six types of nucleotide changes,C→T/G→A, C→A/G→T, C→G/G→C, A→G/T→C,
A→T/T→A, and A→C/T→G. Smohaze carcinogens can cause characteristic mutations (or signature) in the genome.For example, the G to T transversions have been described as a mutational fingerprint of tobacco smoke mutagens in smoking-associated lung cancers, and smokers more frequently show G→T transversions, whereas never-smokers have more G→A transitions45-48. PAHs are the main carcinogens that produce G→T mutations25,49. NNK may induce both G→T and G→A mutations50, and 4-aminobiphenyl and 1,3-butadiene cause the A→T transversions51-53. Other smohaze carcinogens also induce characteristic nucleotide substitutions in the genome54. These mutations were firstly reported in genes such as TP53, RAS,and others25,49,51-53. However, attentions should be paid to the fact that one type of nucleotide changes can be induced by different environmental factors or carcinogens, therefore the so-called “signature” is not stringently specific to one mutagen54,55.
Mutations in specific genes
Husgafvel-Pursiainen et al.56showed that patients who smoke have a three-fold greater risk of TP53 mutations compared to those who do not smoke. Le Calvez et al.57showed that the rate of TP53 mutations increased from 47.5% in never-smokers to 77.4% in active smokers, and the risk of having a TP53 mutation was significantly proportional to the amount of tobacco consumed. KRAS mutations are much more frequent in smokers, in that in active smokers and never-smokers the KRAS mutation rates were 34% and 5%, respectively58,59. BRAF mutations are significantly more frequent in smokers (active or former)60. On the contrary,

Figure 2 Schematic representation of the complicated smohaze-induced lung carcinogenesis.
EGFR mutations and ALK rearrangements are much more frequent in never-smokers compared to active smokers58,59,61,62. Barlesi et al.63further reported significant differences between smokers and never-smokers for mutations in EGFR (4.5% vs. 36.3%), ALK (3.5% vs. 9.7%),KRAS (31.7% vs. 9.6%), BRAF (1.6% vs. 1.8%), and HER2(0.2% vs. 3.8%), respectively (Table 2).
Genomic mutations
Alexandrov et al.64analyzed somatic mutations and DNA methylation in 5,243 cancers of 17 types (including LUAD,LUSC, and SCLC) for which tobacco smoking confers an elevated risk. They showed that smoking is associated with increased mutation burdens of multiple distinct mutational signatures, which contribute to different extents in different cancers. One of these signatures, mainly found in cancers derived from tissues directly exposed to tobacco smoke, is attributable to misreplication of DNA damage caused by tobacco carcinogens. Others likely reflect indirect activation of DNA editing by APOBEC cytidine deaminases and of an endogenous clocklike mutational process. Smoking is associated with limited differences in methylation. Govindon et al.65report the results of whole-genome and transcriptome sequencing of tumor and adjacent normal tissue samples from 17 NSCLCs. They identified 3,726 point mutations and more than 90 small insertions and deletions (indels) in the coding sequence, with an average mutation frequency more than 10-fold higher in smokers than in never-smokers. Deepdigital sequencing revealed diverse clonality patterns in both never-smokers and smokers.
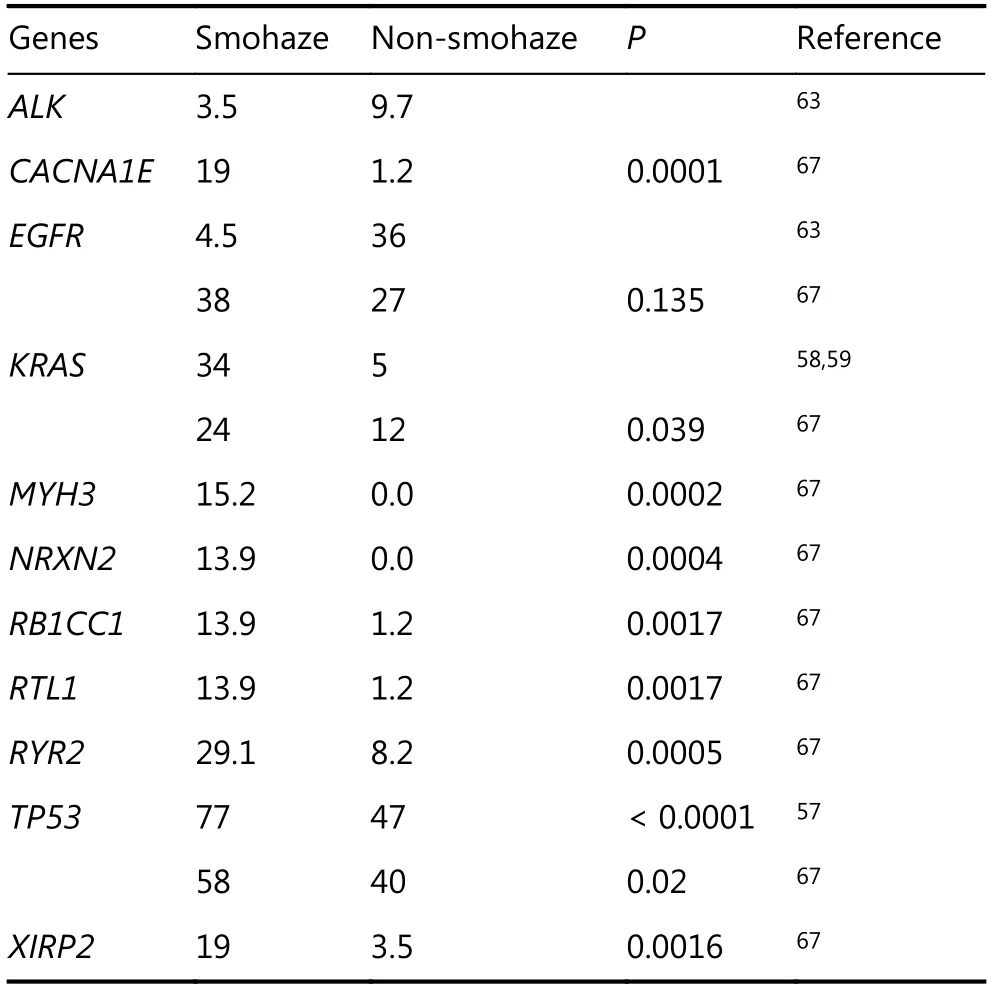
Table 2 Mutation rates of selected genes (10% or more) in NSCLCs associated with smohaze or not
Only one work so far has looked at somatic genomic profiles in association with air pollution66. In that work67, we analyzed the somatic mutations of 164 NSCLCs from XW and control regions (CR) where smoky coal was not used.Whole genome sequencing revealed a mean of 289 somatic exonic mutations per tumor and the C:G→A:T nucleotide substitutions in XW NSCLCs. Exome sequencing of 2010 genes showed that XW and CR NSCLCs had a mean of 68 and 22 mutated genes per tumor, respectively (P<0.0001).We found 167 genes (including CACNA1E, KRAS, MYH3,
NRXN2, RB1CC1, RTL1, RYR2, TP53, XIRP2) which had significantly higher mutation frequencies in HPR than CR patients (Table 2), and mutations in most genes in XW NSCLCs differed from those in CR cases. The mutation rates of 70 genes (e.g., RYR2, MYH3, GPR144, CACNA1E) were associated with patients’ lifetime BaP exposure. This study uncovers the mutation spectrum of air pollution-related lung cancers, and provides evidence for pollution exposuregenomic mutation relationship at a large scale.
Very few studies systematically dissect genomic mutations specifically occurred in “normal” tissues68,69and their roles in lung carcinogenesis. We analyzed the genome of normal lung tissues and paired tumors of patients with LUAD70and LUSC71, and found genomic alterations in the “normal” lung tissues that can be verified by Sanger capillary sequencing.The C:G→T:A transitions, a signature of tobacco carcinogen N-methyl-N-nitro-N-nitrosoguanidine, were the predominant nucleotide changes in counterpart normal controls (CNCs) of LUAD and LUSC. The significance of these variations remains unclear. One possibility was that some of the mutated genes were pro-oncogenes (e.g.,MUC472and CDC2773) or tumor suppressors (e.g.,NCOR174), cells harboring these variations were in a“precancerous” stage, and accumulation of other mutations would result in transformation and development of malignant neoplasms. Secondly, many of the CNC altered genes were associated with immune response, DNA-damage response system, or other important signal pathways, which may facilitate avoiding immune destruction and other hallmarks to facilitate lung cancer70,71. These possibilities warrant further investigation.
Alternative splicing
Alternative splicing, the process by which splice sites are differentially utilized to produce different mRNA isoforms,contributes to oncogenic activation in several types of cancers75,76. Emerging evidence demonstrates that smohaze can induce alternative splicing of some critical genes to facilitate lung carcinogenesis. Weng et al.77showed that of the 117 lung cancer tissue samples analyzed, 31 (26.5%) had splice variants for the MDM2 gene, with 26 samples bearing a splice variant lacking exons 3—11. Significant association was found between the frequency of alternative splicing and the smoking habits of the patients. 44.2% of the smoker patients had alternative MDM2 splice forms versus 16.2% of nonsmokers (P = 0.003). BaP and BPDE induced generation of MDM2 splicing products in H1355 LUAD cells. BPDEinduced MDM2 mRNA alternative splicing in H1355 cells may occur through the PI3K or MAPK pathway. We recently reported a splicing variant of Focal adhesion kinase (FAK),FAK6,7that contains alternatively spliced exons of 18 bp (Box 6) and 21 bp (Box 7) on either side of codon for Y397 in 4 (4.4%) of 91 patients with NSCLC78. Smokers had more FAK abnormalities than non-smokers. In TCGA RNA-seq data, Box 6/7-containing FAK variants were positive in 42(8.3%) of 508 LUADs and 37 (7.4%) of 501 LUSCs, and current smokers had higher expression of Box 6/7 (+) FAK than reformed and never smokers. FAK6,7promoted cell proliferation and migration, and exhibited increased autophosphorylation and was more sensitive to FAK inhibitor compared to wild type FAK78. The effects of smohaze on mRNA splicing and splicing factors warrant further investigation.
Less mutated genes that are critical to environmental lung carcinogenesis
Cancer has been considered as a disease of the genome, and genomic mutations have been shown to be critical to tumorigenesis and served as targets for drug development79.Some genes that are usually wild type also play crucial roles in smohaze-induced lung carcinogenesis.
Aryl hydrocarbon receptor (AhR)
AhR (Figure 3A) is a member of the basic helix—loop—helix—PER— ARNT—SIM (bHLH—PAS) subgroup of the bHLH superfamily of transcription factors. AhR is an environmental sensor integrating immune responses in health and disease80. It can be activated by agonists such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, dioxin) and BaP81, and plays a critical role in endogenous ligand kynurenine-promoted82- and environmental carcinogensinduced tumorigenesis83. A constitutively active AhR promotes hepatocarcinogenesis84and induces stomach tumors85in mice. Shimizu et al.83investigated the response of AhR-deficient mice to BaP, and found that BaP induced subcutaneous tumors in AhR+/+and AhR+/-mice. In contrast, no tumors were apparent in any of the AhRdeficient mice. We recently found that AhR mediated smohaze-induced PD-L1 expression on lung epithelial cells,and deficiency in AhR significantly suppresses BaP-induced lung cancer. AhR inhibitors alpha-naphthoflavone (ANF)and CH223191 exert significant antitumor activity in lung cancer mouse models86. These results indicate that AhR is critical to smohaze-induced lung carcinogenesis, and represents an attractive therapeutic target.
Other genes
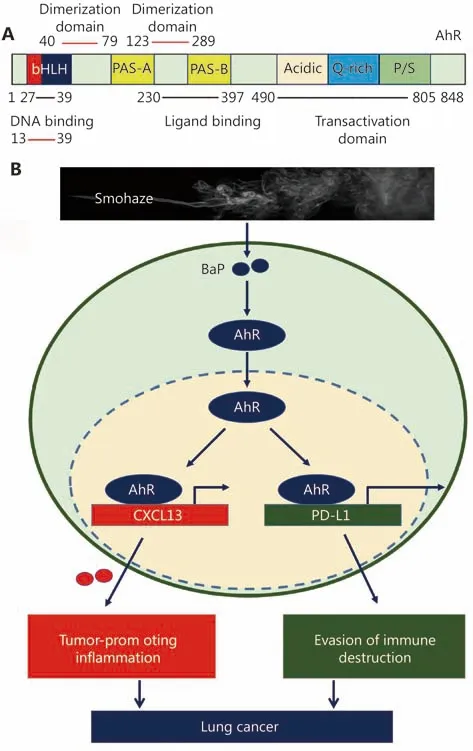
Figure 3 AhR in lung carcinogenesis. (A) Schematic representation of AhR protein. bHLH, basic helix-loop-helix; PAS,period [Per]-aryl hydrocarbon receptor nuclear translocator[ARNT]-single minded [SIM]; P/S, proline (P)/serine (S). (B) AhR mediates smohaze-induced CXCL13 production by PD-L1 expression lung epithelial cells.
Smohaze may perturb the expression of some genes to facilitate lung carcinogenesis. NNK promotes migration and invasion of lung cancer cells through activation of c-Src/PKCi/FAK loop87. Oncoprotein cancerous inhibitor of PP2A (CIP2A) was dramatically elevated in tumor samples compared to paratumor normal tissues of patients with NSCLC88. CIP2A overexpression was associated with patients’ smoking status88, and chronic cigarette smoke exposure induced CIP2A expression in mice89. Silencing CIP2A inhibited the proliferation and clonogenic activity of lung cancer cells. Smohaze may regulate the expression of some genes in an unexpected way. For example, we conducted a large-scale lethality screening in NSCLC cells to silence all the 1530 transcription factors and 696 ubiquitin pathway genes, and found that transcription factor Iroquois Homeobox 5 (IRX5)90and E2 conjugase CDC3490were required for lung cancer cell proliferation. To our surprise,the expression of IRX5 was significantly higher in smoker patients than non-smoker cases, and BaP was able to upregulate IRX5 in lung epithelial cells. Silencing IRX5 significantly inhibited tumor growth in nude mice90. We showed that CDC34 bound EGFR and competed with E3 ligase c-Cbl to inhibit the polyubiquitination and subsequent degradation of EGFR. In EGFR-L858R and EGFRT790M/Del(exon 19)-driven lung cancer in mice,knockdown of CDC34 significantly inhibited tumor formation. CDC34 was elevated in tumor tissues in 67 of 102(65.7%) NSCLCs, and smokers had much higher CDC34 than nonsmokers90. These results indicate that further works should be done to identify critical genes induced or suppressed by smohaze to promote lung carcinogenesis.
Epigenetic abnormalities
DNA methylation
NNK induces DNA methyltransferase 1 (DNMT1)accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients91. An epigenome wide association study was conducted using peripheral-blood DNA of 464 individuals (22 current smokers and 263 exsmokers), and the results suggest the existence of dynamic,reversible site-specific methylation changes in response to cigarette smoking, which may contribute to the extended health risks associated with cigarette smoking92. A metaanalyses of gene methylation and smoking behavior in NSCLCs showed that 7 hypermethylated genes (including
CDKN2A, RASSF1, MGMT, RARB, DAPK, WIF1 and FHIT)were significantly associated with the smoking behavior in NSCLC patients. CDKN2A hypermethylation was significantly associated with cigarette smoking in Japanese,Chinese and Americans, whereas RARB hypermethylation was associated with smoking status in Chinese patients93.Jiang et al.94analyzed genome-wide DNA methylation alterations in XW lung cancers, and obtained a comprehensive dataset of genome-wide CpG island methylation in air pollution-related lung cancers. BaP exposure induced multiple alterations in DNA methylation and in mRNA expressions of DNMTs and ten-11 translocation proteins; these alterations partially occurred in XW lung cancer. BaP-induced DKK2 and EN1 promoter hypermethylation and LPAR2 promoter hypomethylation led to down-regulation and up-regulation of the genes,respectively; the down-regulation of DKK2 and EN1 promoted cellular proliferation. Vitamin C and B6 reduced BaP-induced DNA methylation alterations.
microRNAs (miRNAs)
Schembri et al.95examined whole-genome miRNA expression in bronchial airway epithelium from current and never smokers (n = 20) and found 28 miRNAs to be differentially expressed with the majority being downregulated in smokers. These miRNAs contain potential binding sites for the differentially expressed mRNAs in their 3'-untranslated region (UTR). Among them, miR-218 expression was reduced in primary bronchial epithelium exposed to cigarette smoke condensate. Izzotti et al.96analyzed the expression of 484 miRNAs in the lungs of rats exposed to environmental cigarette smoke (ECS), and found that ECS down-regulated 126 miRNAs (26.0%) at least 2-fold and 24 miRNAs more than 3-fold. The most remarkably down-regulated miRNAs belonged to the families of let-7,miR-10, miR-26, miR-30, miR-34, miR-99, miR-122, miR-123, miR-124, miR-125, miR-140, miR-145, miR-146, miR-191, miR-192, miR-219, miR-222, and miR-223, while miR-294 was up-regulated. They reported a strong parallelism in dysregulation of rodent microRNAs and their human homologues, which are often transcribed from genes localized in fragile sites deleted in lung cancer. Zhang et al.97showed that cigarette smoke upregulates miR-25-3p maturation via N6-methyladenosine and inhibits PHLPP2 to activate AKT, leading to promotion of pancreatic cancer progression. We performed miRNA microarray analysis in NSCLCs from XW or CR, and found 13 down-regulated and 2 up-regulated miRNAs in XW NSCLCs. Among them, miR-144 was one of the most significantly down-regulated miRNAs. The expanded experiments showed that miR-144 was down-regulated in 45/51 (88.2%) XW NSCLCs and 34/54 (63%) CR NSCLCs (P = 0.016). MiR-144 interacted with the oncogene Zeb1 at 2 sites in its 3'-UTR, and a decrease in miR-144 resulted in increased Zeb1 expression and an epithelial mesenchymal transition phenotype. Ectopic expression of miR-144 suppressed NSCLCs in vitro and in vivo by targeting Zeb1. Smohaze can also perturb the expression of other miRNAs to activate oncogenes, suppress tumor suppressors, promote cell proliferation, angiogenesis,and cell cycle, and inhibit apoptosis, to facilitate carcinogenesis98,99.
Long noncoding RNAs (lncRNAs)
lncRNAs are a group of non-coding RNAs consisting of> 200 nucleotides and having no or low translational potential100. Cigarette smoke or BaP can induce the expression of several lncRNAs, such as lncRNA—1 (SCAL1),DQ786227, and LOC728228, in cells101-104. We screened for abnormal lncRNAs in XW lung cancers and reported that XW patients had much more dysregulated lncRNAs than patients from CR. The lncRNA CAR intergenic 10 (CAR10)was up-regulated in 39/62 (62.9%) of the XW patients, which was much higher than in patients from CR (32/86, 37.2%;P = 0.002). PAH compound dibenz (a, h) anthracene (DbA)up-regulated CAR10 by increasing the expression of transcription factor FoxF2. CAR10 bound and stabilized transcription factor Y-box-binding protein 1 (YB-1), leading to up-regulation of EGFR and proliferation of lung cancer cells. Knockdown of CAR10 inhibited cell growth in vitro and tumor growth in vivo105. These results demonstrate the role of lncRNAs in environmental lung carcinogenesis.
Chronic cancer-promoting inflammation
Tobacco smoke induces pulmonary inflammation, which is believed to play a role in progressive lung destruction in COPD106,107. Tobacco smoke induces production of cytokines and chemokines, and chronic inflammation may contribute to tumor initiation/promotion through the production of reactive oxygen and nitrogen species that contribute to DNA damage and induction of oncogenic mutations106,108,109. Takahashi et al.110demonstrated that repetitive exposure to tobacco smoke promotes tumor development both in carcinogen-treated mice and in transgenic mice undergoing sporadic K-ras activation in lung epithelial cells. Tumor promotion is due to induction of inflammation that results in enhanced pneumocyte proliferation and is abrogated by IKKβ ablation in myeloid cells or inactivation of JNK1110. We systematically screened for clinically relevant inflammatory factors that are critical for carcinogenesis, and reported that a chemokine CCL20 was significantly up-regulated by NNK. In 78/173 (45.1%)patients the expression of CCL20 was higher in tumor samples than their adjacent normal lung tissues; CCL20 was up-regulated in 48/92 (52.2%) smoker and 29/78 (37.2%)non-smoker patients (P = 0.05), and high CCL20 was associated with poor prognosis. Anti-inflammation drug dexamethasone inhibited NNK-induced CCL20 production and suppressed lung cancer in vitro and in vivo111. We screened for abnormal inflammatory factors in NSCLCs from XW and CR, and found that a chemokine CXCL13 was overexpressed in 63/70 (90%) of XW NSCLCs and 44/71(62%) of smoker and 27/60 (45%) of non-smoker CR patients. CXCL13 overexpression was associated with the XW region and cigarette smoke. The smohaze carcinogen BaP induced AhR-mediated CXCL13 production in lung epithelial cells and in mice prior to development of detectable lung cancer (Figure 3B). Deficiency in Cxcl13 or its receptor,Cxcr5, attenuated BaP-induced lung cancer in mice,demonstrating CXCL13’s critical role in PAH-induced lung carcinogenesis112. In a nested case-control study (n = 526 lung cancer patients and n = 592 control subjects) measuring serum levels of 77 inflammation markers, CXCL13 and Creactive protein (CRP), CCL22, and IL-1RA provided good separation in 10-year lung cancer cumulative risks among former smokers and current smokers even after adjustment for smoking113. Lung inflammation caused by tobacco smoke exposure also converted disseminated, dormant cancer cells to aggressively growing metastases114.
Immune escape
Whether tobacco carcinogens confer the exposed cells immune escape to initiate carcinogenesis, and why smoker patients response better to immunotherapies than nonsmokers14,15,115, remain poorly understood. We reported that cigarette smoke and carcinogen BaP induced PD-L1 expression on lung epithelial cells in vitro and in vivo, which is mediated by AhR (Figure 3B). Anti-PD-L1 antibody or deficiency in AhR significantly suppresses BaP-induced lung cancer. In 37 patients treated with anti-PD-1 antibody pembrolizumab, 13/16 (81.3%) patients who achieve partial response or stable disease express high levels, whereas 12/16(75%) patients with progression disease exhibit low levels, of AhR in tumor tissues. AhR inhibitors exert significant antitumor activity and synergize with anti-PD-L1 antibody in lung cancer mouse models. These results demonstrate that tobacco smoke induces lung epithelial cells escape from adaptive immunity to promote tumorigenesis, and AhR predicts responses to immunotherapy and represent an attractive therapeutic target. The expression of CTLA-4 was analyzed in 909 NSCLC patients, and the results showed that CTLA-4 expression was significantly higher in LUSC and current/former smokers116. Cigarette smoke also induces lung inflammation and formation of neutrophil extracellular traps, which awaken dormant cancer cells by the neutrophil elastase and matrix metalloproteinase 9114. A recent study showed that carcinogenesis in the lung involves a dynamic co-evolution of pre-invasive bronchial cells and the immune response/immune escape through immune checkpoints and suppressive interleukins from high-grade pre-invasive lesions117.
The anti-lung cancer strategies
To tame lung cancer, WHO20and many countries have launched measures to monitor tobacco use and prevention policies, protect people from tobacco use, offer help to quit tobacco use, warn about the dangers of tobacco, enforce bans on tobacco advertising, promotion and sponsorship, and raise taxes on tobacco. China118and other countries have been tackling the health effects of air pollution. Tremendous efforts have been made to develop preventive and therapeutic approaches. However, the cancer incidence is still high and the 5-year overall survival rate of lung cancer remains dismally low.
Rethinking of the current anti-lung cancer strategies may uncover the following limitations for further improvement.First, smohaze-induced lung carcinogenesis remains to be elucidated. Though numerous studies have been performed to dissect environmental tumorigenesis, many of the works were conducted at cellular and animal models, the clinical relevance of the results should be further tested. Second, no druggable target for smohaze-induced lung cancer was identified and no drug was developed to target smohazeinduced lung carcinogenesis. This represents a major limitation of lung cancer studies in the past 5 decades. Third,while great efforts have been made to characterize genomic mutations, those critical genes that rarely mutate should be unveiled. Thousands of lung cancer genomes have been sequenced in the past decade and a large amount of somatic abnormalities have been reported, but only a few mutated genes have been shown to be the “driver” mutations,suggesting that some less mutated genes should play critical roles in smohaze-induced lung carcinogenesis. AhR represents one of this kind of critical genes, and other critical genes should be uncovered by systematic studies. Of course,the less mutated genes usually play important roles in physiological settings, targeting these genes may possibly induce side effects in the patients. Hence, efforts should be made to scrutinize the requirement of the genes by cancerous and normal tissues, and molecules that are more critical to tumor cells than normal cells could be appropriate targets exemplified by proteasome in multiple myeloma119. Fourth,the current single target-oriented treatment regimens will eventually failed due to development of drug resistance.Inhibition of EGFR gain-of-function mutations marks a revolution in the history of lung cancer treatment, and while resistance occurred the second, third, and even fourth generations of EGFR inhibitors were developed120,121, though drug resistance may develop again. This single target-based passive strategy could be improved by combinatory treatment regimens comprised of different components targeting different targets, exemplified by the traditional Chinese medicine formulae that usually contain compounds of “Jun” (Emperor), “Chen” (Minister), “Zuo” (Assistant),and “Shi” (Delivering Servant) to reach synergistic anticancer effects122.
Perspectives
Up to 90% of cancer cases are caused by environmental factors or lifestyle123. The fact that > 90% of lung cancer deaths are caused by smohaze clearly demonstrates the smohaze-induced lung carcinogenesis as the key to develop effective preventive and therapeutic strategies to tame lung cancer, given that the hazardous health effects of smohaze will not be avoidable for billions of people in the near future.The discoveries that deficiency in AhR significantly inhibited BaP-induced lung86and skin83cancers indicate that inhibition of AhR may exert beneficial effects in prevention and treatment of lung cancer. Moreover, deficiency in CXCL13112or suppression of PD-L186, both of which are AhR target genes, inhibited BaP-induced lung cancer, further confirming the critical roles of AhR in smohaze-induced lung carcinogenesis. With uncovering of more and more critical molecules in smohaze-induced lung carcinogenesis, more targets will be provided to develop more effective preventive and therapeutic approaches. Based on these targets, vaccines and anti-lung cancer formulae can be developed, and early diagnosis and early treatment can be achieved. I believe that these efforts will eventually result in conquering of lung cancer in the future.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (Grant No.2016YFC0905501), the National Natural Science Funds for Distinguished Young Scholar (Grant No. 81425025), the Key Project of the National Natural Science Foundation of China(Grant No. 81830093), the CAMS Innovation Fund for Medical Sciences (Grant No. CIFMS; 2019-I2M-1-003), and the National Natural Science Foundation of China (Grant No. 81672765).
Conflict of interest statement
No potential conflicts of interest are disclosed.
 Cancer Biology & Medicine2019年4期
Cancer Biology & Medicine2019年4期
- Cancer Biology & Medicine的其它文章
- Interpretation of breast cancer screening guideline for Chinese women
- Breast cancer screening guideline for Chinese women
- Erratum to Simultaneous inhibition of PI3Kα and CDK4/6 synergistically suppresses KRAS-mutated non-small cell lung cancer
- The correlation and overlaps between PD-L1 expression and classical genomic aberrations in Chinese lung adenocarcinoma patients: a single center case series
- Nomogram based on albumin-bilirubin grade to predict outcome of the patients with hepatitis C virus-related hepatocellular carcinoma after microwave ablation
- Omics-based integrated analysis identified ATRX as a biomarker associated with glioma diagnosis and prognosis