
壳聚糖亚微粒稳定的Pickering乳液制备pH响应微胶囊
2019-02-13 07:58王静康
天津大学学报(自然科学与工程技术版) 2019年4期
张 欣,谢 闯, ,陈 峰,王静康,
(1.天津大学化工学院,天津 300072;2.天津化学化工协同创新中心,天津 300072;3.天津药业研究院有限公司,天津 300000)
环境响应载药微胶囊可以根据环境条件控制药物释放速率,使药物在不同环境有针对性释放[1].聚乳酸-羟基乙酸共聚物(PLGA)是制备控制药物释放系统最常见的材料[2].由PLGA材料制备的球形微胶囊可以精准控制药物释放长达数十天[3],具有良好的生物相容性和生物降解性.
常见的制备 PLGA微胶囊的方法包括通过 o/w乳液或者w/o/w乳液[4]的溶剂挥发法.微胶囊粒径及分布对于微粒降解和药物释放有很重要的影响.通过控制微胶囊平均粒径和粒度分布可以使生产过程有更好的重现性[5].以乳液为模板制备的微胶囊,乳液的稳定性、粒度及粒度分布直接影响载药微胶囊的质量.普通乳液需要用到较大量的表面活性剂,且乳液滴的稳定性不佳,易聚并分层,这些缺点会对后续微胶囊的制备产生不利影响.Pickering乳液不使用表面活性剂,其乳液稳定剂可以采用亚微米级的固体颗粒,毒性低,且该乳液体系为热力学稳定的体系,稳定性佳[6],可作为很好的微胶囊模板.
壳聚糖(chitosan,CS)作为一种天然多聚糖,兼具生物可降解性和生物相容性,无生物毒性,是最常见的生物医学材料之一.CS与三聚磷酸钠(sodium tripolyphosphate,TPP)通过离子凝胶法制备 CS亚微米颗粒,反应温和且易调控,是使用最多的 CS颗粒制备方法[7].得到的 CS亚微米颗粒(CS-TPP 亚微米颗粒)可以有效稳定 Pickering乳液.在酸性环境,羧甲基纤维素钠(sodium carboxymethyl cellulose,CMC)是一种阴离子天然聚合物.CS中的自由氨基与羧甲基纤维素钠中的羧基可以发生离子间相互作用,形成聚电解质复合物.同时微胶囊的外层包覆一层 CMC,可以使药物从内部到达壳层的跨越距离增大.使微胶囊在酸性介质中稳定,防止药物突释[8],本文将 CMC包覆在 PLGA微胶囊的外层,通过CMC和阳离子聚合物CS-TPP 亚微米颗粒之间的相互作用形成聚电解质复合物.
本文设计制备了一种新的 pH响应性载药微胶囊.以离子凝胶法制备的 CS-TPP 亚微米颗粒为稳定剂,制备o/w型Pickering乳液;以Pickering乳液作为模板,通过溶剂挥发法制备CS-TPP 亚微米颗粒包覆的 PLAG 微胶囊(PLGA/CS-TPP),并在外层包覆CMC,最终得到CMC/CS-TPP 亚微米颗粒包覆的PLGA 微胶囊(PLGA/CS-TPP/CMC);并以布洛芬(ibuprofen,IBU)为模型药物,研究了载药微胶囊在两个不同 pH值的溶液,模拟胃液(simulated gastric fluid,SGF,pH=1.2的盐酸缓冲溶液)和模拟肠液(simulated intestinal fluid,SIF,pH=6.8 的磷酸盐缓冲溶液)[9-10]中药物的体外释放行为.
1 实验部分
1.1 试剂
聚乳酸-羟基乙酸共聚物(PLGA,LA/GA=50∶50,标示黏度 0.41 dl·g-1),济南岱罡生物工程有限公司;冰醋酸(GR,36%,),天津市光复精细化工研究所;二氯甲烷(AR),天津市科密欧化学试剂有限公司;IBU 标准品(分析标准品),索莱宝;IBU(纯度≥99%),萨恩化学技术(上海)有限公司;CS(脱乙酰度>90%,,40万~200万),嘉兴思诚化工;羧甲基纤维素钠(黏度:5,000~15,000,mPa·s,USP 级),麦克林;乙酸乙酯(AR)和三聚磷酸钠(AR),天津市江天化工技术有限公司;氢氧化钠(AR),天津恒山化工科技有限公司;盐酸(AR),天津市江天化工技术有限公司;FITC异硫氰酸荧光素(BR),美国 Sigma-Aldruch公司.
1.2 CS-TPP亚微米颗粒制备
采用修改的离子凝胶法制备 CS-TPP 亚微米颗粒[7].配制不同质量分数 CS醋酸溶液(0.05%,0.10%,0.20%,0.30%),其中CS与醋酸质量比为2∶3.以 0.10%,为例.首先,称取 0.4,g CS粉末,量取10,mL 6%,醋酸水溶液.向醋酸溶液中加入 390,mL超纯水,然后分次加入 0.4,g CS粉末,磁力搅拌过夜,得到分散均一的 CS醋酸水溶液.向 CS水溶液滴加10,mL的TPP水溶液,使CS与TPP的质量比为 5∶1.使用 0.1,mol/L HCl或者 NaOH将溶液 pH值调至4.0,得到淡蓝色CS-TPP 亚微米颗粒悬浮液.
1.3 CS-TPP亚微米颗粒稳定的Pickering乳液制备
将75,mg PLGA溶于1.5,mL二氯甲烷和2,mL乙酸乙酯中,作为油相,将油相滴入 60,mL CS-TPP亚微米颗粒悬浮液.调节磁力搅拌速度,制备出 o/w型Pickering乳液.
1.4 空白微胶囊(PLGA/CS-TPP)制备
通过溶剂挥发法制备 PLGA/CS-TPP.将第 1.3节制得o/w型Pickering乳液移至200,mL烧杯中,倒入100,mL去离子水,室温搅拌6,h,固化得到空白微胶囊.10,000,r/min离心分离 6,min,水洗 3次,收集得到微胶囊.
1.5 包覆CMC的空白微胶囊(PLGA/CS-TPP/CMC)
制备
首先将0.1,g CMC粉末溶解在热的去离子水中,形成CMC溶液,然后将第1.4节所得湿微胶囊转入CMC溶液中,搅拌 40,min.10,000,r/min离心分离6,min,用去离子水冲洗,室温下放置72,h干燥.
1.6 载药微胶囊制备
Pickering 乳液溶剂挥发法制备载药微胶囊过程示意如图1所示.制备载药微胶囊IBU@PLGA/ CSTPP和IBU@PLGA/CS-TPP/CMC时,将一定量药物IBU和 PLGA一起溶解于二氯甲烷和乙酸乙酯中,作为油相,其他步骤与空白微胶囊制备步骤相同.
1.7 微胶囊载药及体外释放实验
1.7.1 载药量的测定
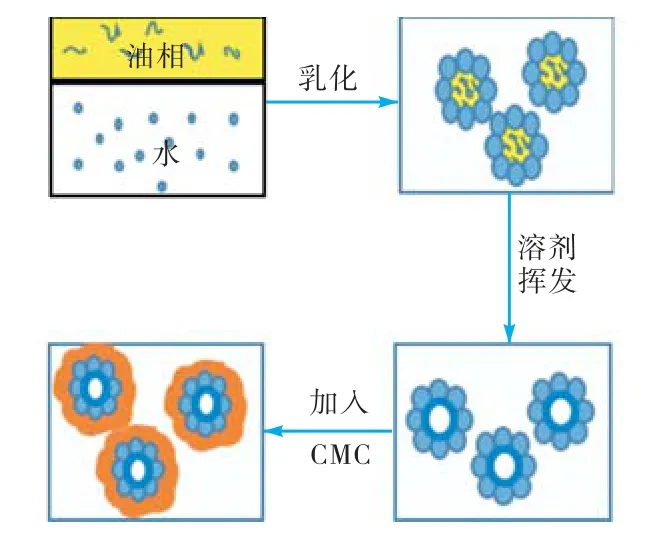
图1 Pickering乳液溶剂挥发法制备微胶囊示意Fig.1 Schematic illustration of the preparation of microcapsules based on solvent evaporation of Pickering emulsion droplets
IBU的载药量通过紫外-可见分光光度计(Hitachi,U-3010)来测量[11].精密称取 20,mg载药微胶囊,溶解于0.5,mL二氯甲烷中,使用无水乙醇定容于 50,mL容量瓶中.10,000,r/min离心 20,min,上清液用滤膜过滤,测得微胶囊中药物质量.
载药量(%,质量分数)的计算式为

式中:m1为微胶囊中布洛芬质量;m2为载药微胶囊总质量.
1.7.2 体外缓释测定
测定 37,℃ IBU从微胶囊向体外释放的累计释放率与时间的关系.采用紫外-可见分光光度计测定药物含量.测定两个不同pH值的溶液中药物的体外释放(模拟胃液(SGF,pH=1.2)和模拟肠液(SIF,pH=6.8))[2].每组实验各称取 25,mg载药微胶囊分散在20,mL缓冲溶液中,然后转入透析袋.将透析袋分别浸没入 180,mL缓冲溶液中,置于 37,℃恒温水浴箱中振荡.在一定时间间隔,取 3,mL透析袋外的清液,进行吸光度测试,并同时迅速补充 3,mL新鲜的缓冲溶液.通过计算得到IBU的体外累计释放率.
1.8 物化性能表征
乳液的形貌通过光学显微镜(Optec,BK6000)进行观察.CS、CS-TPP 亚微米颗粒、PLGA、IBU、空白微胶囊(PLGA/CS-TPP)及载药微胶囊(IBU@PLGA/CS-TPP和IBU@PLGA/CS-TPP/CMC)的红外光谱则通过傅里叶红外光谱仪(Broker公司,TENSOR 27)测得.微胶囊的表面形貌通过光学显微镜、激光共聚焦显微镜(美国 PerkingElmer公司,UltraView Vox,激发光波长 488,nm)和场发射扫描电子显微镜(SEM,Hitachi,TM3030)进行观察.使用Nano ZS型纳米粒度(英国马尔文公司,Nano ZS)测定 CS-TPP亚微米颗粒粒径及 zeta电势.利用接触角测量仪(美国科诺,SL200KS),采用静滴法测量CS-TPP颗粒干燥后的膜层浸在油水两相的接触角,表征润湿性.测量时,将涂有固体颗粒的盖玻片浸没在水相中,使用接触角测量仪测量,通过仪器上的自动进样注射器将油相液(二氯甲烷∶乙酸乙酯=3∶4(体积比))滴在样品上,连续拍摄水滴在样品片上的形貌,从而测量出滴在盖玻片上的油滴与固体层之间的角度.
2 结果与讨论
2.1 CS-TPP亚微米颗粒与Pickering乳液的表征
所制备的CS-TPP亚微米颗粒的红外光谱如图2所示.在纯CS红外谱图中,3,356,cm-1是CS的O—H 伸缩振动峰,1,645,cm-1、1,590,cm-1和 1,317,cm-1分别是酰胺Ⅰ带、酰胺Ⅱ带和酰胺Ⅲ带吸收峰[10].与之相比,1,645,cm-1和 1,590,cm-1处的吸收峰在 CSTPP 亚微米颗粒中分别移动到 1,625,cm-1和1,532,cm-1,表明 CS的氨基和 TPP发生了明显的作用.这表明CS分子中带正电的质子化氨基与TPP分子中带负电的磷酸根相互作用、产生交联,从而获得了CS-TPP 亚微米颗粒[7].
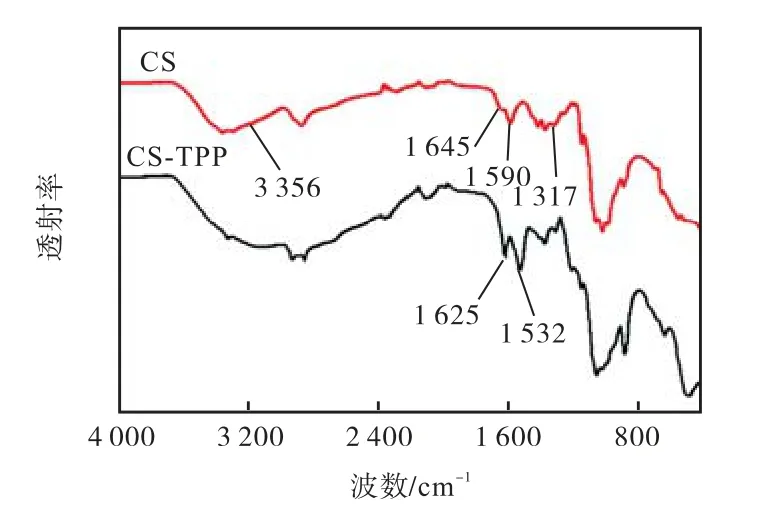
图2 CS-TPP亚微米颗粒的红外谱图Fig.2 FT-IR spectra of CS-TPP submicron particles
CS-TPP 亚微米颗粒作为 Pickering乳液的稳定剂,其粒度大小、zeta电势以及表面润湿性直接影响Pickering乳液的稳定性.本文所得 CS-TPP 亚微米颗粒制备的膜层在水环境中与油滴的接触角为 134°(如图 3所示),颗粒表面显亲水性,接触角角度大,适合用来稳定乳液.对于 Pickering乳液,当颗粒表面较大部分浸在连续相中时,颗粒表面优先被连续相润湿,提高了液体界面凸液面处力学屏障的有效性[12].因此,基于亲水性 CS-TPP亚微米颗粒,可制备o/w型Pickering乳液.

图3 CS-TPP 亚微米颗粒在油水界面的接触角分析Fig.3 Contact angle test between CS-TPP particles and oil-water interface
本文在CS和TPP质量比为5∶1的条件下,考察了4种CS质量分数所得的CS-TPP颗粒.结果表明,CS-TPP颗粒的粒径随着 CS质量分数增大而增大(如图 4所示).CS水溶液质量分数从 0.05%,增加到 0.30%,时,CS-TPP 颗粒平均粒径从 600,nm 增加到37.8,μm.在酸性条件下,溶液中的CS分子处于带正电质子化氨基的静电排斥与分子间氢键相互吸引的平衡[12-13].带负电TPP的加入降低了静电排斥,导致 CS与 TPP的交联,形成 CS-TPP颗粒.随着 CS质量分数的增大,CS分子相互靠近,分子间的交联增多,导致粒径变大.但当 CS质量分数接近 0.20%,时,颗粒粒径增大为微米级,不利于后续Pickering乳液的稳定.本文选择 0.10%,CS质量分数制备的 CSTPP亚微米级颗粒,平均粒径650,nm,zeta电势为+34.4,mV,适合作为后续Pickering乳液的稳定剂.

图4 CS质量分数对CS-TPP 颗粒平均粒径的影响Fig.4 Effect of CS mass ratio on mean size of CS-TPP particles
对制备的 CS-TPP亚微米级颗粒稳定的 Pickering乳液,采用光学显微镜对其形貌进行表征,如图5所示.采用稀释法[14]鉴别出制备的乳液为 o/w乳液.液滴形状为球形,平均粒径为 266,μm.制备的o/w乳液稳定性可以保持一个月以上,为后面的溶剂挥发过程提供了稳定的模板.

图5 Pickering乳液的显微镜图片Fig.5 Optical microscopy image of Pickering emulsions
2.2 Pickering乳液和微胶囊形貌及粒径分布
图 6给出了包覆 CMC前后微胶囊的显微镜图像及其粒径分布.如图6(a)所示,液滴的平均粒径为266,mm.对经溶剂挥发并固化后所得的微胶囊,采用光学显微镜和 SEM 对其形貌及表面进行表征.可以看出包覆 CMC前的 PLGA/CS-TPP球形度好、形状规则、粒径分布均匀.对显微镜图像中 200个微胶囊的粒径进行统计,得到 PLGA/CS-TPP微胶囊的平均粒径为 154,μm,变异系数 15.1%,;包覆 CMC 后,PLGA/CS-TPP/CMC(图 6(b))保持了良好的球形度和粒径分布,粒径略有增加,平均粒径约为 160,μm,变异系数 16.7%,.从图 7的 SEM 照片可以看出,包覆CMC前的PLGA/CS-TPP表面有一层褶皱的膜层(图 7(a)和(b)),应是由于 CH2Cl2蒸发,乳液体积减小,表面颗粒壳层收缩所致.包覆 CMC后,PLGA/CS-TPP/CMC 表面(图 7(c)和(d))较包覆前更平整,表明CMC成功包覆.
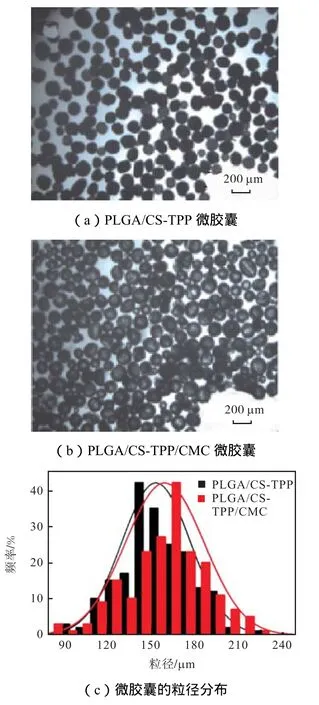
图6 微胶囊的显微镜图像及粒径分布Fig.6 Optical microscopy images and the size distributions of microcapsules

图7 载药微胶囊的SEM照片Fig.7 SEM images of drug-loaded microcapsules
为进一步验证所制微胶囊的表面为 CS-TPP颗粒,采用荧光染料FITC标记CS,并通过激光共聚焦显微镜对标记后的微胶囊进行表征,结果如图 8所示.可以看出微胶囊的外表面有一圈明显的荧光物质(图 8(a)),而微胶囊内核几乎没有荧光强度(图8(b)).由于FITC特异地与CS-TPP亚微米颗粒上的氨基结合[14],这也确认了在微胶囊表面存在一层CSTPP亚微米颗粒保护层.
2.3 微胶囊红外光谱分析

图8 微胶囊的激光共聚焦图像Fig.8 Confocal microscopy image of microcapsules
为验证药物 IBU是否包封在 PLGA微胶囊中,对载药微胶囊进行了红外表征,并将其与空白微胶囊以及纯PLGA和IBU的红外谱图进行了对比,如图9所示.纯 PLGA(图 9(a))在 1,746,cm-1处特有的C=O吸收特征峰也出现在空白微胶囊 PLGA/CSTPP(图 9(b))和载药微胶囊(图 9(c)和 9(d))中,表明PLGA在微胶囊中的存在.与空白胶囊相比,载药后的IBU@PLGA/CS-TPP和IBU@PLGA/CS-TPP/CMC的谱图中多出了 IBU的特征峰(1,508,cm-1处C=C(苯环上)伸缩振动峰和 779,cm-1处=C—H(苯环上)伸缩振动峰)[15-17],表明 IBU 已经被成功地包裹在载药微胶囊中.包覆 CMC后的 IBU@PLGA/CS-TPP/CMC比包覆前的IBU@PLGA/ CS-TPP多出了纯 CMC(图 9(f))的特征峰(1,595,cm-1为 C=O吸收峰[18]),表明CMC成功地包覆在微胶囊表面.
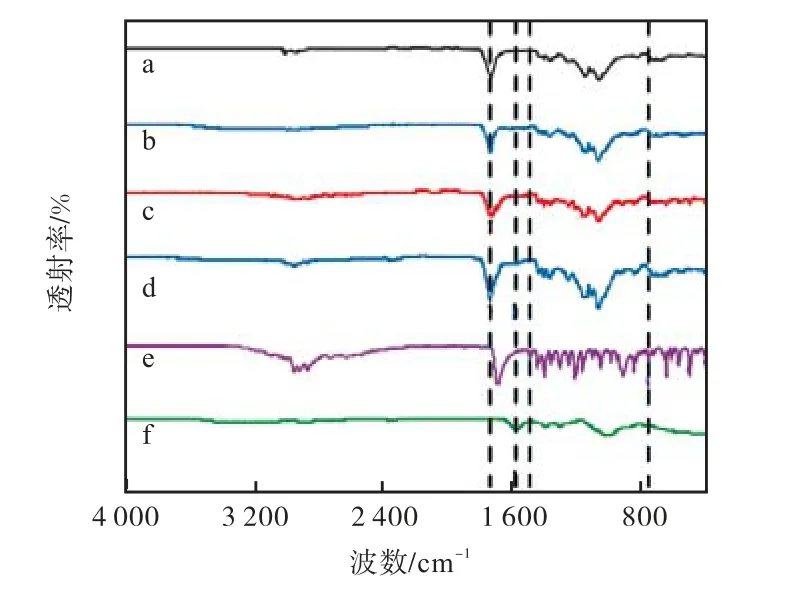
图9 载药前后微囊红外光谱对比Fig.9 Comparison of FT-IR spectra of microcapsules before and after loading drug
2.4 载药微胶囊的体外释放行为评价
对所得 IBU@PLGA/CS-TPP/CMC微胶囊的载药量测试结果表明,其载药量为 24%,.包覆CMC前后的载药微胶囊在SIF和SGF中的释放曲线如图10所示.结果表明,没有包覆CMC的IBU@PLGA/CSTPP微胶囊在SGF中的的释放速度低于SIF,表现出了一定的 pH响应性,但释放速率差异不明显.IBU在SGF中溶解度较在SIF中低是造成这种释放率差异的原因之一.而包覆 CMC后的 IBU@PLGA/CSTPP/CMC微胶囊,在 SGF中释放速度远低于其在SIF中,8,h内平均释放速度相差 12倍,表现出优良的pH响应性.CMC所引起的这种pH响应性变化的原因可能有:①CMC在表面的结合使得微胶囊表面更加致密,同时在物理上起到稳定微胶囊结构的作用,阻碍药物的向外扩散;②在酸性溶液(SGF)中,IBU@PLGA/CS-TPP/CMC表面 CMC的-COO-基团质子化,增加了疏水性,而IBU@PLGA/CS-TPP表面的CS颗粒层更加亲水,因此,在SGF中CMC进一步阻碍了 IBU的扩散;③在 SIF中,CMC层的—COO-电离,亲水性增强,溶液加速通过壳层[8],导致 IBU@PLGA/CS-TPP/CMC中的药物释放速率提高.

图10 IBU@PLGA/CS-TPP/CMC和IBU@PLGA/CS-TPP载药微胶囊体外缓释曲线Fig.10 Cumulative release profiles of IBU@PLGA/CSTPP/CMC and IBU@ PLGA/CS-TPP microcapsules
3 结 论
基于 CS-TPP 亚微米颗粒稳定的 Pickering乳液,设计制备了pH响应的载药微胶囊.
(1) 制备CS-TPP 亚微米颗粒时,CS和TPP质量比为 5∶1,CS质量分数为 0.10%,条件下可制备650,nm 的 CS-TPP 亚微米颗粒,适于用来稳定Pickering乳液.
(2) 基于 Pickering乳液可以方便有效地获得亚微米颗粒稳定的载药微胶囊,在制备过程中微胶囊结构不易被破坏;亚微米颗粒可有效减少常规乳化方法中的表面活性剂的使用.
(3) 引入 CMC可提高载药微胶囊稳定性的同时有效增强其 pH响应性;包覆CMC后的载药微胶囊在SIF中的释放速率是在SGF中的约12倍,表现出良好的pH响应性.
[1] Cong Y,Chen K,Zhou S.Synthesis of pH and UV dual-responsive microcapsules with high loading capacity and their application in self-healing hydrophobic coat[J].Journal of Materials Chemistry A,2015,3(37):19093-19099.
[2] Fredenberg S,Wahlgren M,Reslow M,et al.The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review[J].International Journal of Pharmaceutics,2011,415(1/2):34-52.
[3] Makino K,Nakajima T,Shikamura M,et al.Efficient intra cellular delivery of rifampicin to alveolar macrophages using rifampicin-loaded PLGA microspheres:Effects of molecular weight and composition of PLGA on release of rifampicin[J].Colloids and Surfaces B:Biointerfaces,2004,36(1):35-42.
[4] 黄慧玲,李 益,王 玥,等.5-FU-PLGA复乳微球的制备及体外释放[J].化学工业与工程,2016,33(1):76-81.
Huang Huiling,Li Yi,Wang Yue,et al.Preparation and in vitro release of 5-FU-PLGA double emulsion microsphere[J].Chemical Industry and Engineering,2016,33(1):76-81(in Chinese).
[5] 王 彦,王靖涛.微流控技术制备聚酰胺微胶囊的工艺研究[J].化学工业与工程,2017:1-6,doi:10.13353/j.issn.1004.9533.20161118.
Wang Yan,Wang Jingtao.Preparation of polyamide microcapsules based on microfluidics[J].Chemical Industry and Engineering,2017:1-6,doi:10.13353/j.issn.1004.9533.20161118(in Chinese).
[6] Wu J,Ma G H.Recent studies of Pickering emulsions:Particles make the difference[J].Small, 2016,12(34):4633-4648.
[7] Hu Bing,Pan Chenliang,Sun Yi,et al.Optimization of fabrication parameter to produce chitosantripolyphosphate nanoparticles for delivery of tea catechins[J].Journal of Agricultural and Food Chemistry,2008,56(16):7451-7458.
[8] Rafi A A,Mahkam M.Preparation of magnetic pH-sensitive microcapsules with an alginate base as colon specific drug delivery systems through an entirely green route[J].RSC Advances,2015,5(6):4628-4638.
[9] 国家药典委员会.中国人民共和国药典[M].4部.北京:中国医药科技出版社,2015.
Chinese Pharmacopoeia Commission.Chinese Pharmacopoeia[M].Part 4.Beijing:China Medical Science Press,2015(in Chinese).
[10] Ma G.Microencapsulation of protein drugs for drug delivery:Strategy,preparation,and applications[J].Journal of Conterolled Release,2014,193:324-340.
[11] 杨建红,杜予民,覃彩芹.红外光谱与核磁共振波谱在甲壳素结构研究中的应用[J].分析科学学报,2003,19(3):282-287.
Yang Jianhong,Du Yumin,Qin Caiqin.Applications of infrared spectroscopy and nuclear magnetic resonance spectroscopy in the studies of the structure of chitin and chitosan[J].Journal of Analytical Science,2003,19(3):282-287(in Chinese).
[12] Aveyard R,Binks B P,Clint J H.Emulsions stabilised solely by colloidal particles[J].Advances in Colloid and Interface Science,2003,100(2):503-546.
[13] Fan W,Yan W,Xu Z,et al.Formation mechanism of monodisperse,low molecular weight chitosan nanoparticles by ionic gelation technique[J].Colloids and Surfaces B:Biointerfaces,2012,90(1):21-27.
[14] Liu H,Gu X,Hu M,et al.Facile fabrication of nanocomposite microcapsules by combining layer-by-layer self-assembly and Pickering emulsion templating[J].RSC Advances,2014,4(32):16751-16758.
[15] Wei Z,Wang C,Liu H,et al.Facile fabrication of biocompatible PLGA drug-carrying microspheres by O/W pickering emulsions[J].Colloids and Surfaces B:Biointerfaces,2012,91(1):97-105.
[16] Wang J,Law W,Chen L.Fabrication of monodisperse drug-loaded poly(lactic-co-glycolic acid)-chitosan coreshell nanocomposites via pickering emulsionuntitled[J].Composites Part B:Engineering,2017,121:99-107.
[17] Chen F,Zhu Y.Chitosan enclosed mesoporous silica nano particles as drug nano-carriers:Sensitive response to the narrow pH range[J].Microporous and Mesoporous Materials,2012,150:83-89.
[18] 林粤顺,周红军,周新华,等.2,4-D/CMC-g-AM/SA/FK微球的制备及其缓释性能[J].化学研究与应用,2015,27(4):456-461.
Lin Yueshun,Zhou Hongjun,Zhou Xinhua,et al.Preparation and slow-release performance of 2,4-D/sodium carboxymethyl cellulose grafted acrylamide/sodium alginate/feather keratin microsphere[J].Chemical Research and Application,2015,27(4):456-461(in Chinese).
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26
中国饲料(2021年17期)2021-11-02
食品安全导刊(2021年21期)2021-08-30
食品安全导刊(2021年21期)2021-08-30
石油沥青(2021年1期)2021-04-13
石油沥青(2021年6期)2021-02-10
上海建材(2021年1期)2021-01-12
中国药学药品知识仓库(2021年17期)2021-01-11
中国现代医生(2018年30期)2018-12-06
优雅(2016年2期)2016-06-03
