
相关基因多态性的沙坦类药物药代动力学研究进展
2019-02-11 11:05宋忠金周静李宁赵娣陈西敬
药学研究 2019年12期
宋忠金,周静,李宁,赵娣,陈西敬
(中国药科大学基础医学与临床药学学院临床药物代谢动力学研究室,江苏 南京 211198)
作为一种血管紧张素Ⅱ受体特异性拮抗剂,沙坦类药物有着长效、高效的降压效果[1],但此类药物个体间药动学行为存在较大差异[2]。影响沙坦类药物体内处置过程的代谢酶和转运体主要包括P-糖蛋白(P-gp),多耐药基因1(MDR1)[3]、细胞色素氧化酶(CYP2C9)[4]、多药耐药相关蛋白(MRP2)[5]、有机阴离子转运多肽(OATP)和葡萄糖醛酸转移酶(UGT)等[6-7]。这些大分子基因多态性所导致的编码蛋白的活性改变,可能与沙坦类药物的个体差异存在一定的联系。针对目前常用的沙坦类药物,本文对沙坦类药物基因组学研究的进展和展望进行综述。
1 P-gp 与沙坦类药物
1.1 P-gp基因型分布 胃肠道功能是影响口服药物吸收的第一步。P-gp作为一种跨膜转运蛋白,由人类MDR1编码[8],其在多个组织中表达,尤其是肠道,是许多脂溶性化合物的外排泵,依赖于ATP能量供应在肠上皮细胞中挤压多种底物以降低细胞内药物浓度,保护细胞免受有毒物质的侵害,许多药物都是MDR1的底物[9]。MDR1基因位于染色体7q21.1上,由28个外显子(exon)组成,共编码大约1 300个氨基酸, MDR1是高度多态性的,迄今为止,至少有50个单核苷酸多态性(SNPs)已被发现[10]。这些MDR1遗传变异会造成许多药物个体间的药代动力学和药效学差异[11]。在对MDR1众多SNPs的研究中,主要集中在MDR1 exon 12 C1236T,exon 21 G2677T/A和exon 26 C3435T,其中位于26位外显子上的C3435T研究最多,该位点为无义突变,不引起氨基酸的变化,但却会改变与底物的相互作用,可能是因其空间构型不同[12]。C3435T的突变导致P-gp蛋白表达的改变,纯合子CC基因型会上调P-gp的表达[13]。该突变位点在不同种族中也存在较大差异[14-15], 如表1所示,在亚洲人群中,巴基斯坦、印度、中国人群中的基因型频率明显具有差异,而欧美人群与亚洲人之间,差异也较大,这些都会导致不同种族间的用药差异,因此,MDR1 C3435T多态性的阐释对个体化药物治疗具有重要意义。
1.2 P-gp基因多态性对沙坦类药物药代动力学的影响 影响沙坦类药物个体差异的因素很多,作为P-gp的底物,MDR1基因多态性可能是导致其差异的重要原因之一。如表2所示,Yasar等[16]挑选了58名健康土耳其受试者,在排除CYP2C9的干扰下(受试者基因型均为CYP2C9*1/*1),研究MDR1 3435T基因多态性对氯沙坦药动学的影响。结果表明,58名受试者中,CC、CT、TT基因型的频率分别为22.4%、50%、27.6%。单次口服25 mg氯沙坦后,CC、CT、TT 3种基因型受试者体内氯沙坦及其代谢物E3174的平均血药浓度分别为(1.76±0.87 μmol·L-1,2.97±2.49 μmol·L-1)、(1.68±0.84 μmol·L-1,2.53±2.09 μmol·L-1)、(1.80±0.85 μmol·L-1,3.18±2.75 μmol·L-1),根据统计学分析,不同基因型之间血药浓度并无显著性统计差异,提示该突变位点对氯沙坦的药动学行为并无显著影响。
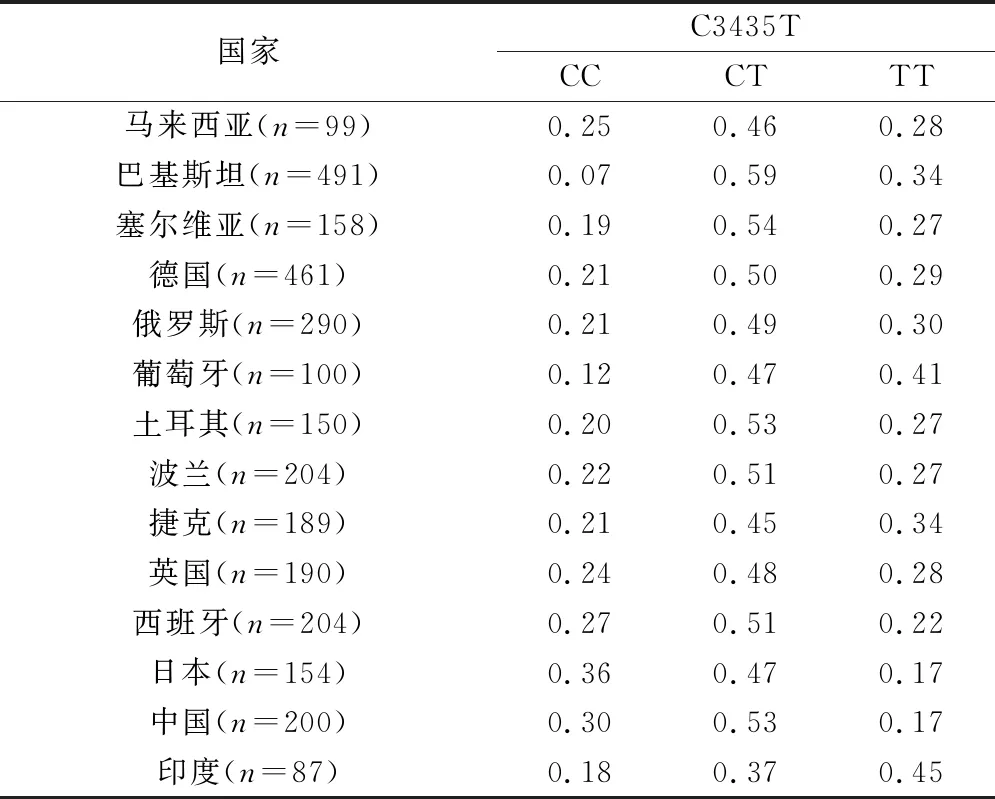
表1 不同种族群体中MDR exon 26 C3435T多态性的基因型和等位基因频率
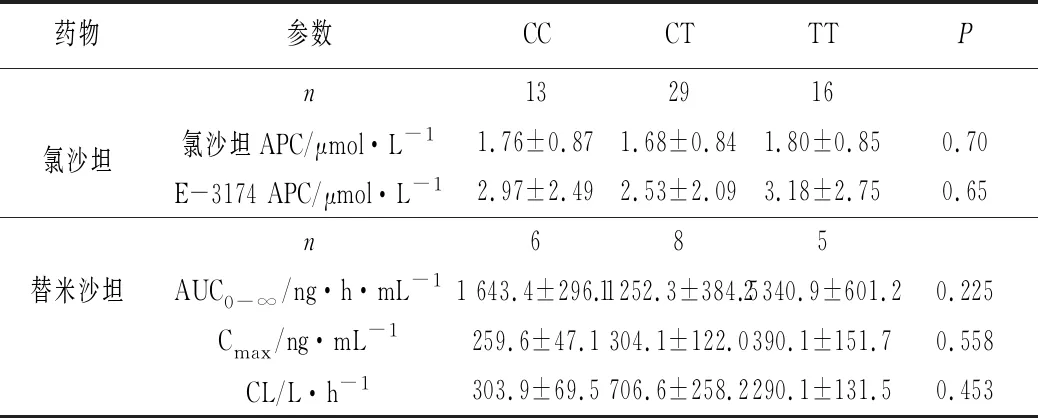
表2 氯沙坦与替米沙坦与MDR1 C3435T单核苷酸多态性的药代参数关系
注:APC-平均血浆浓度
国内关于C3435T基因与沙坦类药物的动力学关系研究较多,范秀珍等[17-18]均研究了MDR1 C3435T 基因多态性对替米沙坦药代动力学参数的影响。结果显示,CC、CT、TT 3种不同基因型在中国人群中的突变频率大约分别为40%、50%、10%。但无论是健康受试者,还是高血压患者,替米沙坦在3种基因型人体内的药代参数差异均无统计学意义。张欣然等[19]则进行了C3435T不同基因型对厄贝沙坦和缬沙坦血药浓度的相关性研究,结果表明稳态血药浓度并没有在3个突变体间显示出具体差异。
虽然C3435T基因突变频率较高,在不同人群中差异较大,并且显著影响P-gp对其底物的转运结果,但综合目前的国内外研究,该基因位点并不影响沙坦类药物的药动行为,因此可以推断,临床用药时可能不需考虑C3435T基因突变的相关性。
2 CYP2C9与沙坦类药物
2.1 CYP2C9基因型研究 CYP2C9是人肝中主要的CYP2C蛋白,是肝药酶家族中重要的一员,占肝微粒体CYP450量的20%,并且介导大约10%的药物的I相代谢。CYP2C9基因位于染色体10q24,有9个负责克隆490个氨基酸残基蛋白质的外显子。目前已发现60多种CYP2C9突变体,研究最多的等位基因是CY92C9*2和CYP2C9*3。CYP2C9*2基因在exon 3 的碱基被替换(430C>T),相应氨基酸序列也发生改变(144Arg>Cys),CYP2C9*3基因exon7 1075位的碱基被替换(1075A>C),导致第359位的亮氨酸被取代为异亮氨酸[20]。国内外均报道CYP2C9*2、*3突变频率较低,Miners等[21]研究显示*2、*3两个等位基因在白种人中的突变频率分别为8%~12.5%,3%~8.5%,在汉族人和蒙古人中CYP2C9*2的突变频率也仅为0.011 0、0.025 3[22]。虽然突变的频率降低,但这些多态性可能影响2C9的活性,可能改变底物的代谢清除速率并引起血液中药物浓度的变化,这可能影响功效并引起不良反应。因此,研究CYP2C9基因多态性对于指导沙坦类药物的临床使用具有重要意义。
2.2 CYP2C9基因多态性对沙坦类药物药动学的影响 大部分沙坦类药物如缬沙坦、氯沙坦、坎地沙坦和厄贝沙坦等在人体内均由CYP2C9代谢[23]。氯沙坦是世界上第一个上市的AT1受体拮抗剂,它通过CYP2C9主要代谢为更强活性的E-3174[24]。研究表明,CYP2C9*2和CYP2C9*3突变可降低CYP2C9对氯沙坦的代谢活性。在分析CYP2C9*3(rs1057910A>C)基因多态性与氯沙坦的药代动力学中,陈露露等[25]研究发现,与AA基因型相比,AC基因型受试者中氯沙坦的药动学参数AUC及Cmax明显升高,存在显著差异,但其代谢产物E-3174的参数没有显著差异(见表3)。值得注意的是,在后来对CYP2C9*2基因多态性进行分析的时候,杨璐等[26]发现,在服用氯沙坦后,相比于CYP2C9*1/*2组,CYP2C9*1/*1组受试者氯沙坦代谢物E-3174的AUC0-24、AUC0-∞、Cmax分别是前者的1.36、1.32和1.64倍,呈现显著性差异,而原药却无明显差别。这表明CYP2C9在氯沙坦的代谢过程中起着至关重要的作用,是其个体差异较大的因素之一。Chen等[27]在对196名高血压患者服用厄贝沙坦后的药动学和药效学研究分析发现,CYP2C9*3基因变异显著改变了中国高血压患者厄贝沙坦治疗后6 h时的血浆浓度和急性舒张压(diastolic blood pressure,DBP)反应。
而在国外的研究中,Choi等[28]在对28名韩国人研究后发现,单次口服150 mg厄贝沙坦后,与CYP2C9*1纯合子受试者相比,CYP2C9*1/*3的Cmax、T1/2与时间曲线下面积AUC分别是前者的1.56、1.38和1.64倍。同时,厄贝沙坦口服清除率降低了39.3%,表明CYP2C9的基因突变可以降低厄贝沙坦的体内代谢。CYP2C9的遗传多态性研究在国内外相对较为普遍,其与沙坦类药物药代动力学之间的关系正逐步被阐明。截至目前,CYP2C9*2、*3位点的突变频率虽然较低,但其却能显著降低相应编码蛋白的活性,减少沙坦类药物的代谢,使其暴露量增加,从而增加不良反应风险,所以在临床实际用药时,应将CYP2C9基因型考虑在内,但目前尚没有文章报道增加沙坦类药物的系统暴露量会对患者产生什么样的具体影响,还需要更多临床试验来证实。
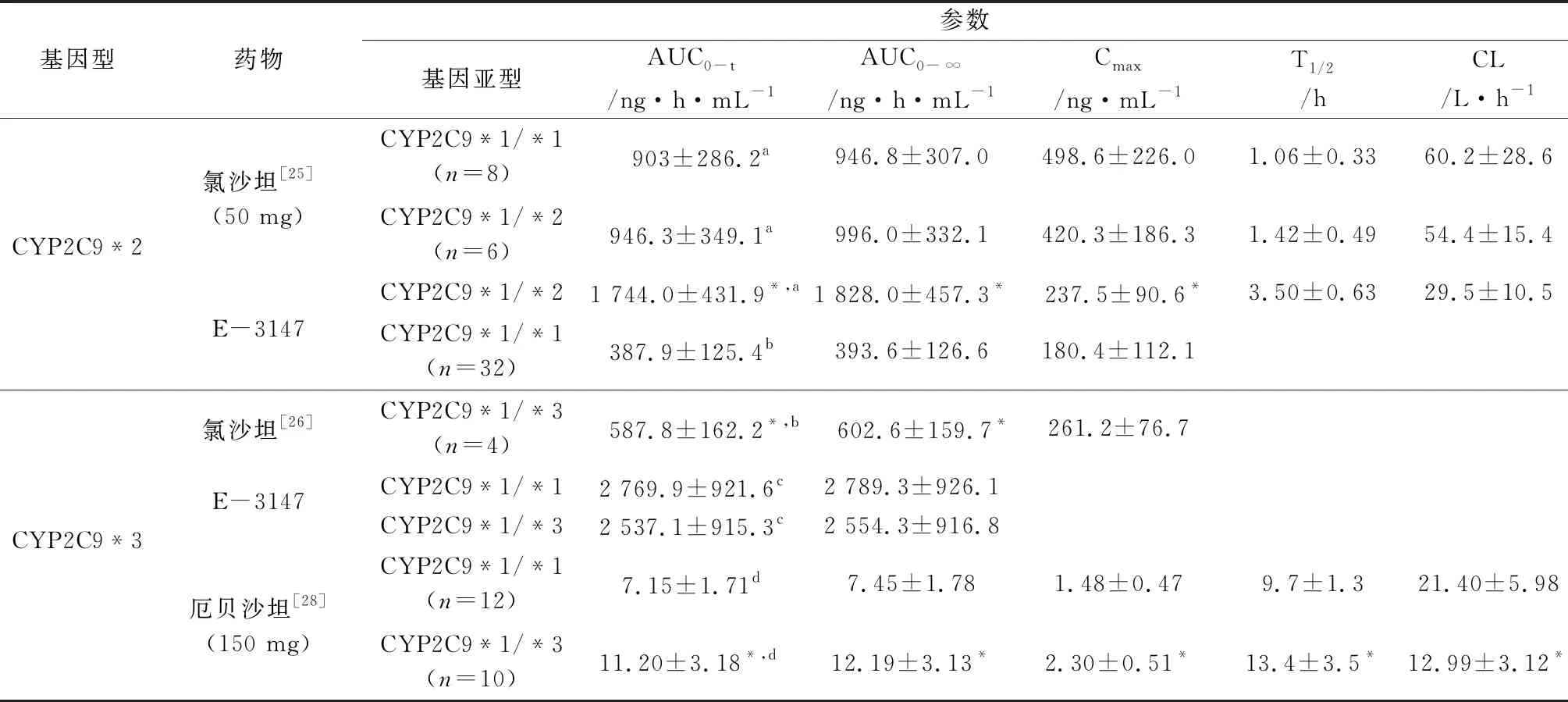
表3 CYP2C9多态性与沙坦类药物药代参数关系
注:a、b、c、d分别代表0~24、0~12、0~48、0~36 h的AUC0~t,与CYP2C9*1/*1比较,*P<0.05
3 OATP1B1与MRP 2与沙坦类药物
OATP1B1/MRP2/BCRP组合代表了在肝脏转运中起重要作用的三大有机阴离子转运体。药物首先通过OATP1B1摄入,然后通过MRP2或BCRP排出,这构成了药物肝脏胆汁排泄的载体转运过程[29]。OATP1B1是一个高变异基因,目前已确认41个非同义突变,最常见的是exon 5上的521T>C(Val172Ala,rs4149506)等位基因突变,导致转运体活性的降低,限制肝细胞摄取药物。此外,388A>G(Asn130Asp,rs2306283)亦是一个发生频率较高的位点[30]。多药耐药相关蛋白2(MRP2)主要分布在肝细胞和肾小管细胞的腔膜中,可以将药物从细胞中排进邻近腔隙,最常见的突变位点是C-24T、G1249A、C3972T,三者大概突变频率为20%、12%、22.5%。其中,只有G1249A可引起氨基酸变化(取代417位的缬氨酸变为异亮氨酸)[31]。田蕾等[29]在研究OATP1B1和 MRP 2对缬沙坦的影响时发现,以23名521TT野生型志愿者为实验对象,在排除OATP1B1 T521C基因多态性的影响后,缬沙坦药动学参数在2个基因组间表现出显著差异。相比于388GG组,388AA-AG组的AUC、Cmax分别高出51%、39%,突变体纯合388GG的AUC与Cmax均显著低于杂合388AA-AG,表明388GG纯合突变导致OATP1B1转运活性升高和肝脏摄取增加,导致血药浓度降低,而MRP 2 -24C>T并未产生影响(见表4)。而Kim等[32]在研究这两转运体对奥美沙坦的药代影响时,却得出了不一样的结论,数据表明ABCC2 -24CC基因组的Cmax和AUC显著低于-24CT组,然而,OATP1B1并未对奥美沙坦的暴露量产生显著影响。两个转运体对缬沙坦和奥美沙坦产生药代参数差异的原因可能是因为另一个肝脏转运体OATP1B3参与,而替米沙坦是OATP1B3的代偿性转运底物,从而抵消了MRP2基因多态性的影响。同时,底物不同、种群不同、样本例数少等等也是产生差异的可能原因,所以需要更多的临床试验,来明确OATP 1B1和MRP 2基因多态性对沙坦类药物的影响。
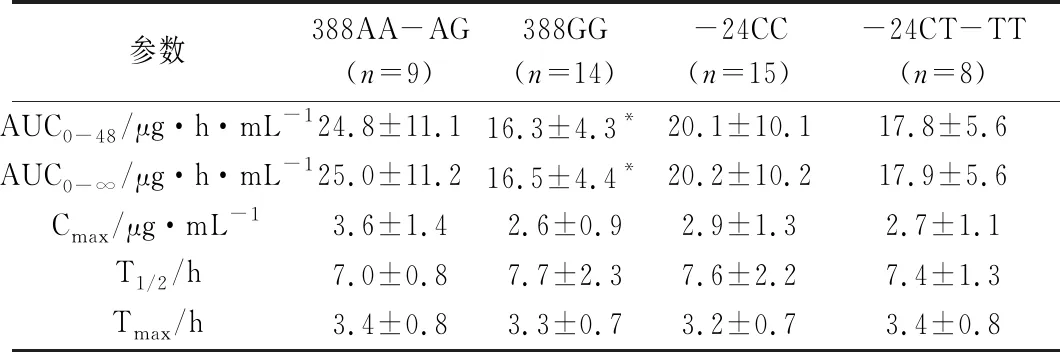
表4 OATP1B1和MRP2基因多态性与缬沙坦药动学参数关系[29]
注:*P<0.05 表明有显著性差异
4 UGT1A3与沙坦类药物
如上所述,肝药酶2C9参与了大多数沙坦类药物的体内代谢,但替米沙坦通过UGT代谢为无活性的葡糖苷酸,这增加水溶性并且易于排泄。目前研究显示,UGT1A3 exon 1共发现6个SNP位点,其中4个(-17A/G,Q6R;-31T/C,W11R;-133C/T,R45W;-140T/C,V47A)可引起氨基酸的改变,另外两个为静默位点(-81G/A和-477A/G)[33]。UGT1A3突变位点在中国人群中发生频率较高,Chen等[34]检测125位受试者的基因,其中T31C、G81A、T104C的频率分别为26.8%、26.8、10.4%。而白种人中这三个基因的突变频率更是分布高达65%、65%、58%[35]。Iieri等[7]在2011年首次考察了33名日本健康男性受试者葡萄糖醛酸转移酶1A3(UGT1A3)基因多态性与替米沙坦药动学之间的关系。实验证明,UGT1A3*1a/*1a携带者Cmax高于UGT1A3*2a携带者(749±430,724±541 ng·mL-1)(P<0.05),UGT1A3*4a携带者Cmax显著高于UGT1A3*1a/*1a携带者(1 421±479 ng·mL-1,749±430 ng·mL-1)(P<0.01)。同时,UGT1A3*1a/*1a、*2a、*4a携带者的AUC0-24h分别为2 969±1 456、1 701±970、5 340±1 168 ng·h·mL-1,差异显著。对于替米沙坦的清除率,UGT1A3*2a、*4a分别为*1a/*1a的2.19和0.55倍。可以看出,携带UGT1A3*2a基因的受试者具有最高的酶活性,体内消除较快。反之,UGT1A3*4a携带者的酶活性较低,消除缓慢。2013年刘容吉[33]考察了中国健康受试者中UGT1A3多态性对替米沙坦药代动力学的影响,在分析了UGT1A3*1/*1、UGT1A3*1/*2及UGT1A3*1*4三个基因型后,结果表明,三组受试者替米沙坦的Cmax、AUC、Vd/F间均有显著性差异,与日本受试者的体内结果基本一致。因此,UGT可能是替米沙坦个体血药浓度差异大的原因之一。
5 展望
沙坦类药物在人体内具有较长半衰期,大多在10 h以上,替米沙坦的半衰期更是长达24 h,除了高效,有良好的耐受性之外,也减少了患者的用药频率。但其个体差异大,临床效果及副作用因人而异,这与遗传因素的影响密不可分。随着基因分析技术的快速发展,不同个体基因型对药物效应的差异关系正在逐渐被阐明。近些年来,随着全球各地研究人员针对沙坦类药物的基因多态性研究,我们对其关联性有了一定的了解。但研究至今,尚未达成共识,形成普遍的指导原则。因此,要推进沙坦类药物的基因组学研究,需要不同地区和种族的研究人员间加强合作,深入探索,制定科学统一的标准和方案,选用灵敏、高效、可靠的检测方法,扩大临床样本量,进行合理的前瞻性设计,开展研究。
猜你喜欢
化工管理(2022年14期)2022-12-02
昆明医科大学学报(2021年6期)2021-07-31
世界最新医学信息文摘(2021年12期)2021-06-09
安徽医科大学学报(2021年4期)2021-04-09
云南化工(2020年11期)2021-01-14
家庭医药·快乐养生(2019年1期)2019-01-30
天津医科大学学报(2015年2期)2015-12-22
药学与临床研究(2015年6期)2015-03-06
中国当代医药(2015年26期)2015-03-01
中国当代医药(2015年16期)2015-03-01
