
农药原药全组分分析杂质结构剖析
2019-01-14 09:21曹立冬吴进龙张宏军李凤敏黄啟良
农药科学与管理 2018年11期
曹立冬,吴进龙,张宏军,李凤敏,黄啟良*
(1.生物性危害因子控制重点实验室/中国农业科学院 植物保护研究所,北京 100193;2.农业农村部农药检定所,北京 100125)
农药原药全组分分析是指针对有效成分、含量达到0.1%及以上的任何杂质以及FAO、WHO或者是各国农药主管部门规定的任何含量水平的相关杂质(即对人类和环境具有明显的毒害,或者对使用作物产生药害,或引起农产品污染,或影响农药产品质量稳定性,或引起其他不良影响的杂质)的定性和定量分析,鉴定出的组分总的质量含量一般须在98%~102%之间[1]。农药登记资料一般需提供至少5个批次具有代表性的工业化生产原药的全组分分析报告,所以原药全组分分析又称5批次全分析。农药全分析资料是进行国内外农药登记及市场开发不可或缺的重要技术资料。对新农药而言,全分析报告是进一步对其安全性进行评价的重要化学依据;对于申请相同产品登记而言,全分析报告是认定其是否与已登记原药“等同”的重要依据。
原药全组分分析最有技术含量且最有挑战性的工作是含量>0.1%的杂质的定性鉴定。原药中的杂质可以比作是原药的“指纹”,是判断原药合成工艺的重要参数,也是相同产品认定的关键要素。作者根据近年来从事原药全分析的经验,结合实例分析,谈谈原药中杂质结构剖析的一些思路和方法。
1 原药中杂质来源
原药中的杂质主要来自于反应原料(原料、原料中杂质和溶剂)、中间体和副反应产物。副反应主要包括降解、异构化、水(溶剂)解、氧化反应、过度取代、重排等反应。在众多来源中,原药合成所用的原料及合成中间体是最容易判断且标准品易得的,原药生产企业基本上都有每步反应的中间体保留且存档。原料中的杂质比较难以推断的,需要对原料进行定性分析从而判断其在原药中的结构。原药中最常见的杂质来自于副反应,包括以上提到的各种类型的反应。因此,快速准确的进行原药中杂质的结构推断,需要具有丰富的有机化学反应理论和实践经验。
2 原药中杂质结构剖析的一般思路
对于原药全分析,首先要进行预实验分析确定含量≥0.1%的杂质个数,一般借助于液相色谱或者气相色谱分析,通过峰面积百分比进行杂质个数的确定。对于液相色谱,有效成分和杂质的色谱响应与所选择的检测波长紧密相关,如果仅仅选择最适合有效成分的波长,则有些杂质可能会因该波长下响应弱,出现峰面积百分比<0.1%或者没有色谱响应。因此,为了准确进行杂质个数的判断,建议选择3D扫描模式进行原药预分析。图1为噻苯隆原药3D扫描模式下的色谱图,可以清晰的看出每个杂质组份在每个波长下的色谱响应情况。
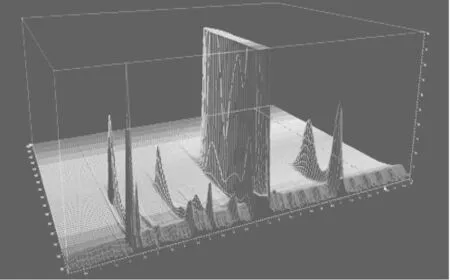
图1 噻苯隆原药色谱图(3D扫描模式)
色谱确定好杂质个数后,进行质谱分析,根据质谱数据结合合成工艺,推断杂质可能的结构。质谱分析也可以进行杂质数量的确定,有的杂质色谱响应很弱,却具有很强的质谱响应,这点也需要在全分析过程中引起足够的重视。对于结构比较确定的杂质,如果合成步骤简单就通过合成获取杂质标准品,或者直接进行购买商品化标准品。对于含量较高,且与有效成分及其它杂质分离度较高杂质,根据质谱信息难以推断出合理的化学结构,通过液相制备色谱进行富集,然后结构鉴定。对于含量很低的杂质,可以先通过重结晶原药,提高杂质含量,母液进行液相色谱制备。杂质标准品获得后,通过与原药中杂质色谱保留时间(液相还需要比对紫外吸收光谱图)和质谱数据的比对,进行杂质结构的定性确定。
3 原药中杂质结构剖析实例分析
3.1 利用核磁裂分和偶合常数进行异构体结构鉴定 对于同分异构体,仅仅通过质谱数据难以准确进行结构判断,利用核磁裂分和偶合常数可进行异构体的结构鉴定。氰氟草酯原药通过质谱解析,发现存在一个同分异构体杂质,推断可能的结构,进行定向合成。由于亲核取代反应的选择性,得到2个同分异构体1和2(图2)。化合物1和2由于极性接近,难以通过常规的柱层析分离,我们借助于液相制备色谱进行分离,然后分别进行核磁共振氢谱(1H NMR)测定。借助于核磁裂分和偶合常数进行异构体结构鉴定(图3),通过色谱保留时间比对分析,确定氰氟草酯原药中存在的同分异构体杂质为化合物2。
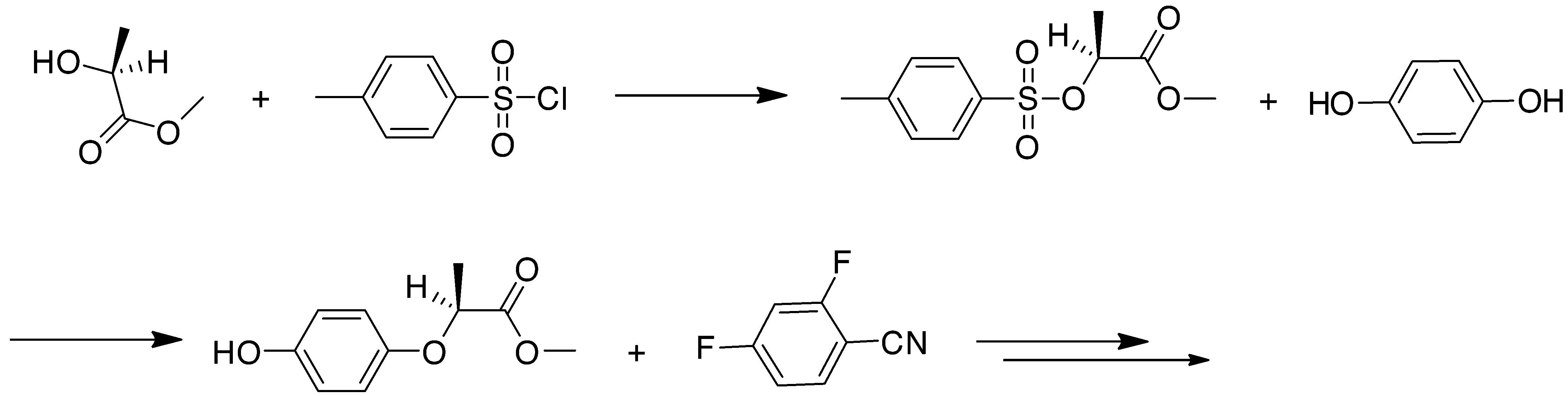
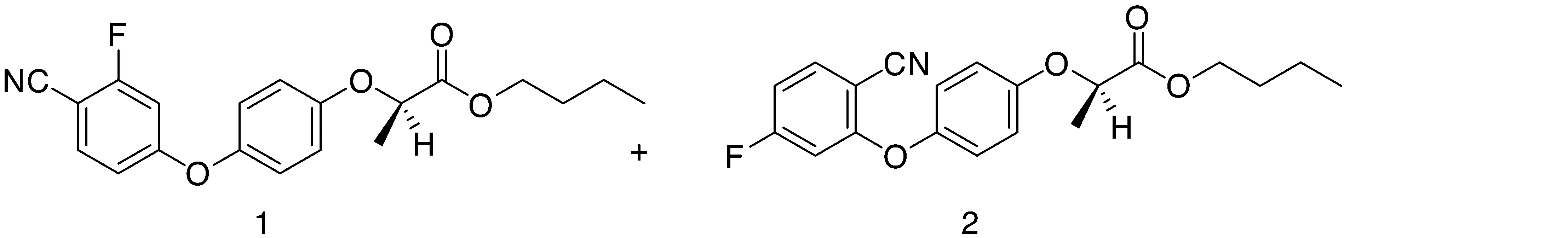
图2 氰氟草酯同分异构体杂质合成路线
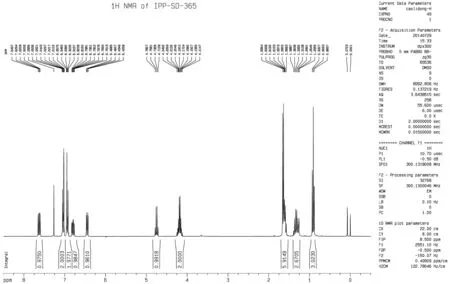
图3 氰氟草酯同分异构体杂质21H NMR谱图
在氟磺胺草醚原药全分析中,通过质谱解析,发现存在一个双硝基化的杂质,由于硝基可能存在芳环不同的位置,给结构鉴定带来了困难。我们根据合成工艺及可能的反应位点,推断可能的双硝基化杂质,进行定向合成,得到2个双硝基化杂质3和4(图
4),然后分别进行核磁共振氢谱(1H NMR)测定。借助于核磁裂分和偶合常数进行异构体结构鉴定(图5),通过色谱保留时间比对分析,确定氟磺胺草醚原药中存在的双硝基化杂质为化合物3。
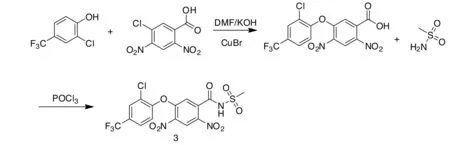
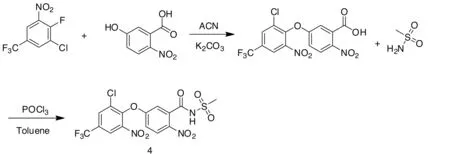
图4 氟磺胺草醚原药双硝基化杂质合成路线
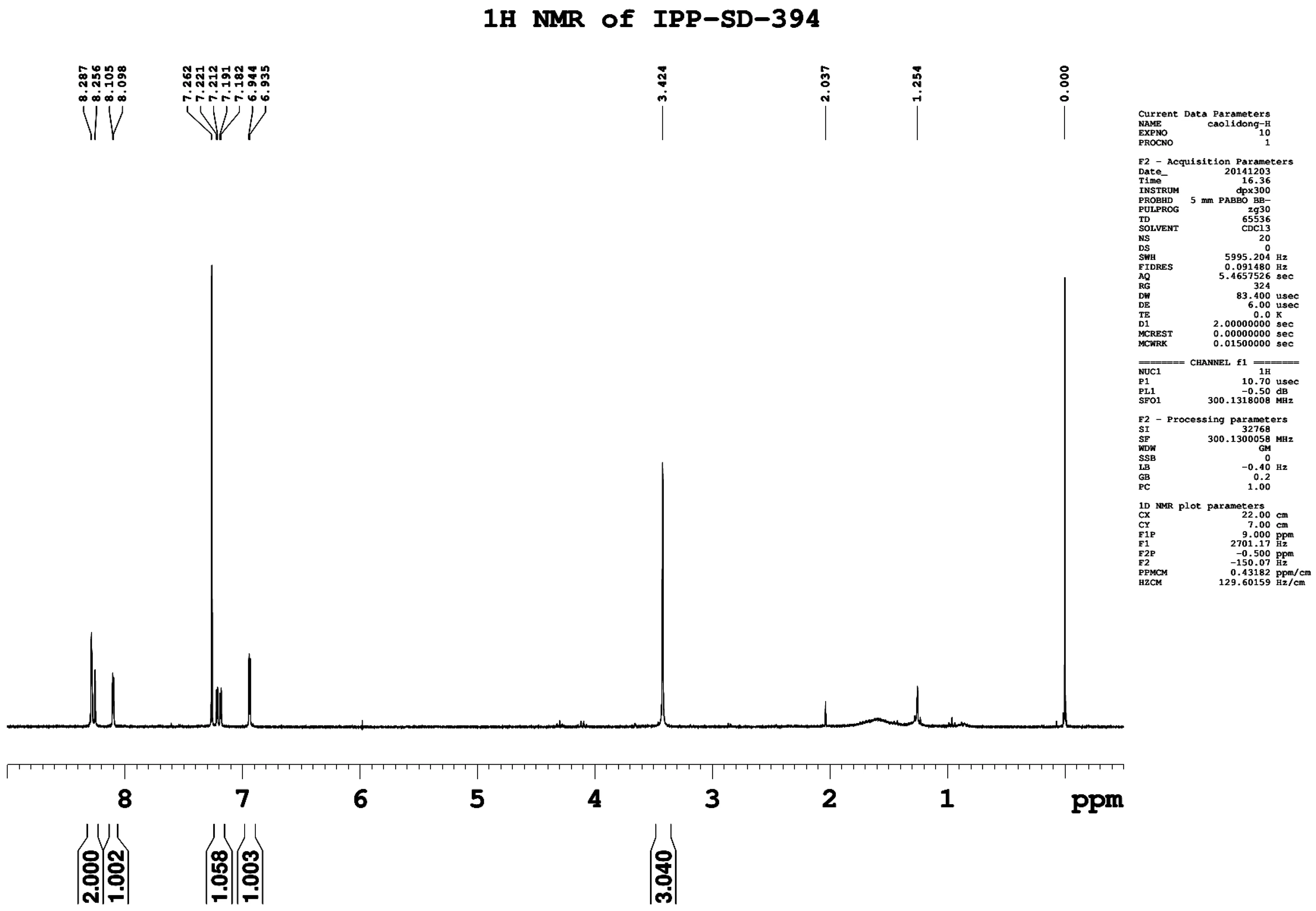
图5 氟磺胺草醚原药双硝基化杂质1H NMR谱图
3.2 水解副反应杂质鉴定 原药有效成分合成工艺中经常会用到水溶液,并且原药储存过程中也很容易接触到空气中的水分子,从而发生水解反应。水解产物是常见的一种副反应杂质,该类杂质也非常容易制备,原药在适当的条件进行水解反应可大量制备水解产物。以氰氟虫腙和螺虫乙酯为例,水解产物5和6可以方便的通过一步反应制备(图6),反应后经过简单的过滤和重结晶即可得到高纯度水解产物。
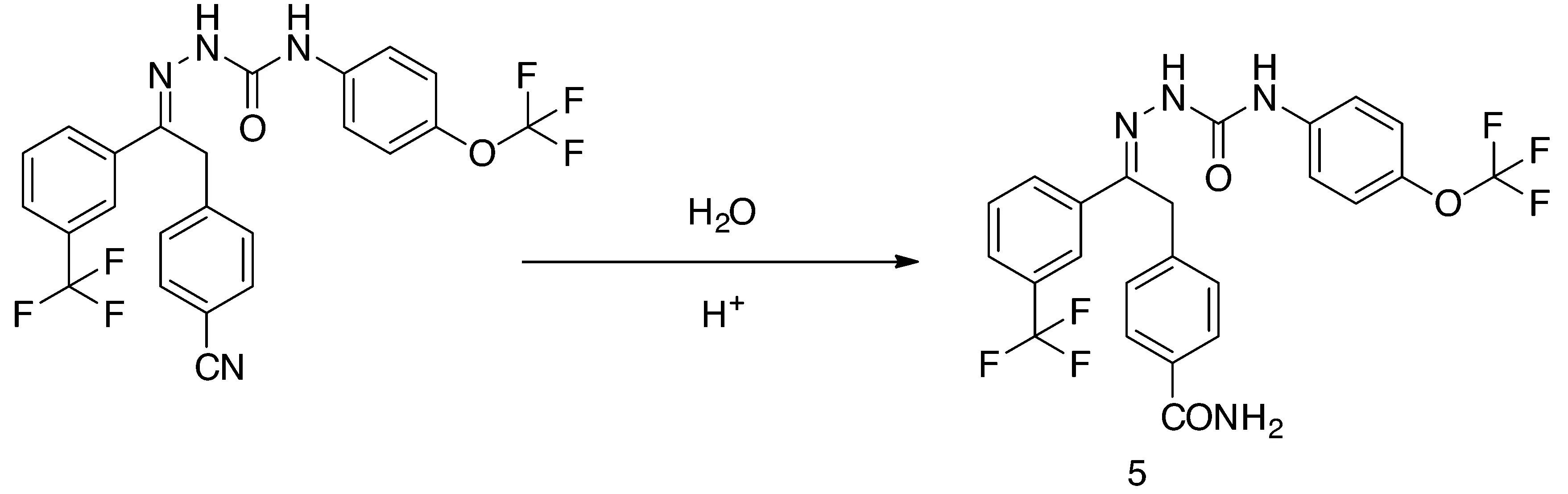
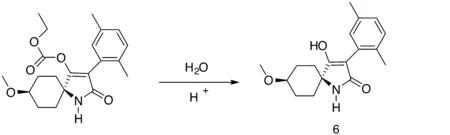
图6 氰氟虫腙和螺虫乙酯水解产物5和6合成路线图
3.3 有机溶剂参与副反应杂质鉴定 原药在合成工艺过程会经常用到有机溶剂,原药全分析也会用到有机溶剂溶解。如果有机溶剂参与反应,就会发生溶剂解反应,从而带来新的杂质。在做异菌脲全分析过程中,配制异菌脲溶液最初选择的是甲醇,发现有新的色谱峰出现,且随着时间的延长,该色谱峰峰面积逐渐增加,通过液质分析,发现该化合物为异菌脲的同分异构体7(图7),甲醇作为亲核试剂进攻异菌脲导致开环异构化(图8)[2]。

图7 异菌脲甲醇异构化反应
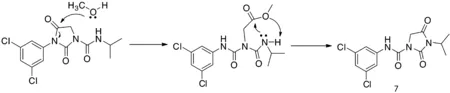
图8 异菌脲甲醇异构化反应可能的机理
吡唑醚菌酯原药中经常出现质荷比为435的杂质,结合原药合成工艺,我们推断是原药合成最后一步的中间体与反应溶剂1,2-二氯乙烷进行亲核取代反应带来的杂质8(图9)。
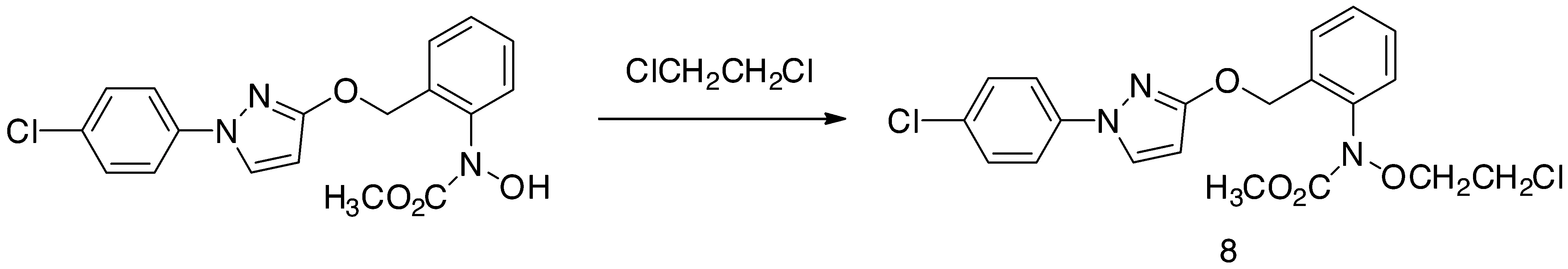
图9 吡唑醚菌酯原药中质荷比为435杂质生成途径
3.4 多(少)卤(氟、氯、溴)代副反应杂质鉴定 卤素(氟、氯、溴)广泛存在于农药分子中,在卤代反应过程中经常出现多(少)卤代杂质出现,卤素原子的个数可以通过质谱数据中[M+4]:[M+2]:[M]的丰度比判断。在苯嗪草酮原药全分析中,通过质谱解析,发现多一个氯原子的杂质,结合合成工艺,我们推断可能的杂质为苯环上氯原子对位取代,并设计了该杂质(化合物9)的合成路线(图10)。非常幸运的是,我们合成的杂质正好是原药中存在的杂质,该杂质的结构也经过了1H NMR确证(图11)。
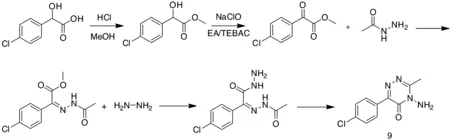
图10 苯嗪草酮原药中氯代杂质合成路线
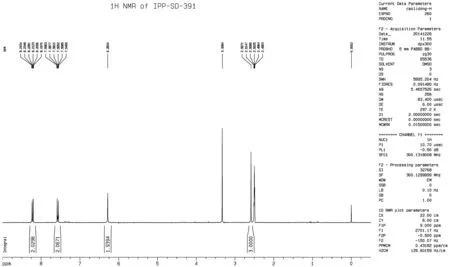
图11 苯嗪草酮原药中氯代杂质1H NMR谱图
吡唑醚菌酯原药中经常出现质荷比为465的杂质,根据同位素相对丰度,推断该杂质为吡唑醚菌酯多一个溴原子。结合原药合成工艺,我们推断可能是吡唑醚菌酯的吡唑环与溴发生了溴代反应,生成溴代杂质10(图12),为进一步验证该杂质的结构,我们一步合成了该溴代杂质,并与原药中的杂质进行了比对确认。

图12 吡唑醚菌酯原药中溴代杂质合成路线
3.5 不完全(过度)氧化(还原)反应杂质鉴定 氧化还原反应是农药合成工艺中非常常见的反应,不完全(过度)氧化(还原)会带来副产物。吡唑醚菌酯原药中出现质荷比为610的杂质,按照正常的杂质推断思路,非常难以解析。仔细分析原药合成工艺,发现中间体存在一步硝基还原反应,理想的中间体产物应该为羟胺,如果硝基还原不完全则会生成亚硝基,羟胺与亚硝胺进行加成反应并消除一分子水,则生成质荷比为610的杂质11(图13)。乙虫腈由硫醚氧化得到,过度氧化会生成杂质12(图14)。
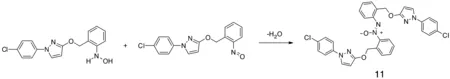
图13 吡唑醚菌酯原药中质荷比为610杂质生成途径
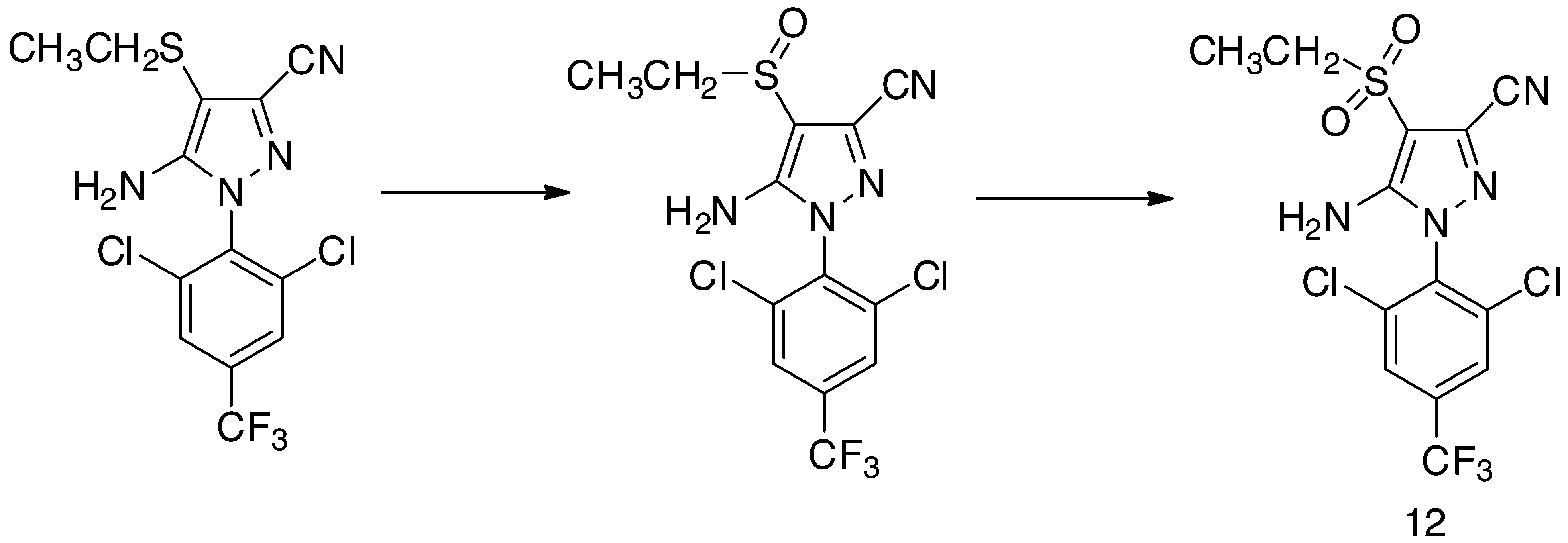
图14 乙虫腈过度氧化杂质生成途径
3.6 重排反应杂质鉴定 化合物在某些特定的条件下会发生重排异构化或者重排消除反应,从而导致杂质的生成。鱼藤酮环外双键会发生重排异构化反应得到热力学更稳定的环内双键异构体13(图15)。啶嘧磺隆原药会发生重排消除反应得到杂质14,进一步发生水解反应可得到杂质15(图16)。
3.7 特殊反应杂质鉴定 有机化学反应类型众多,有一些不太常见,我们可称之为特殊反应。比如在稻瘟灵原药全分析过程中,发现了一个结构高度对称的杂质16,结合合成工艺,我们推断有硫代二烯酮中间体生成,发生[2+2]环加成反应即可得到杂质16(图17)。该杂质的结构进一步经过了1H NMR确证(图18)。
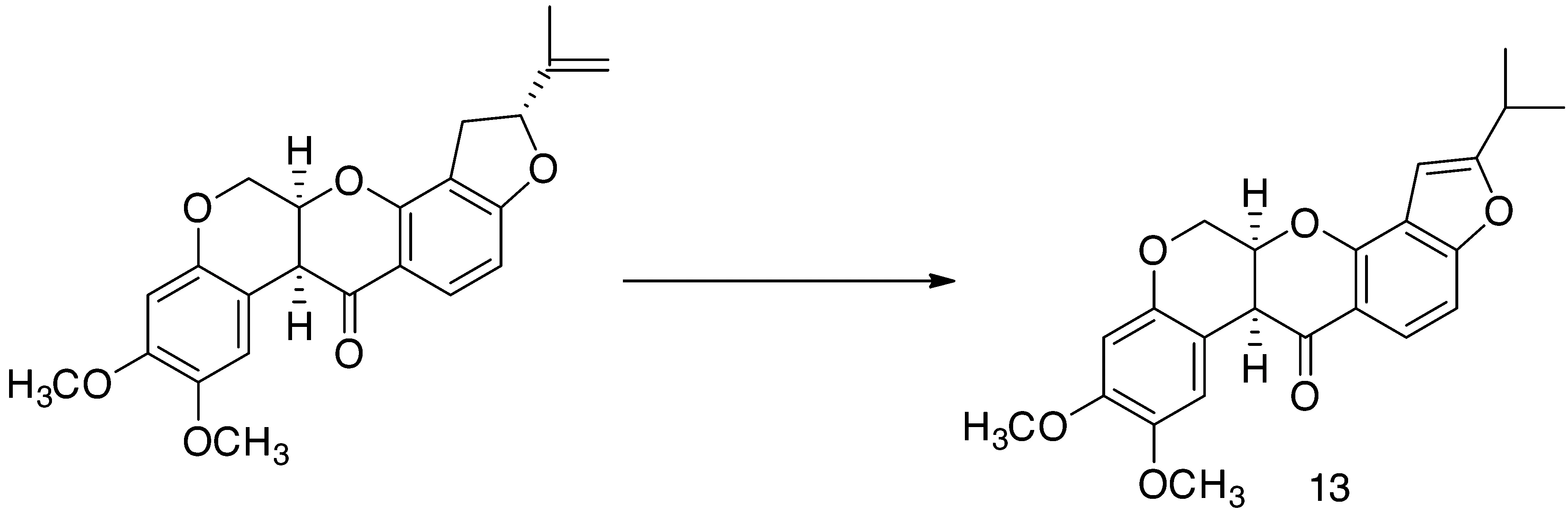
图15 鱼藤酮环外双键异构化
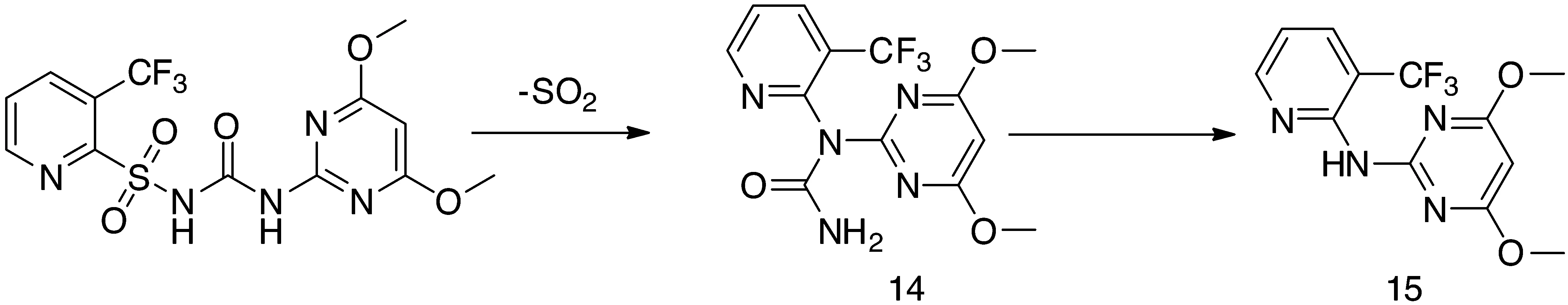
图16 啶嘧磺隆重排消除反应途径
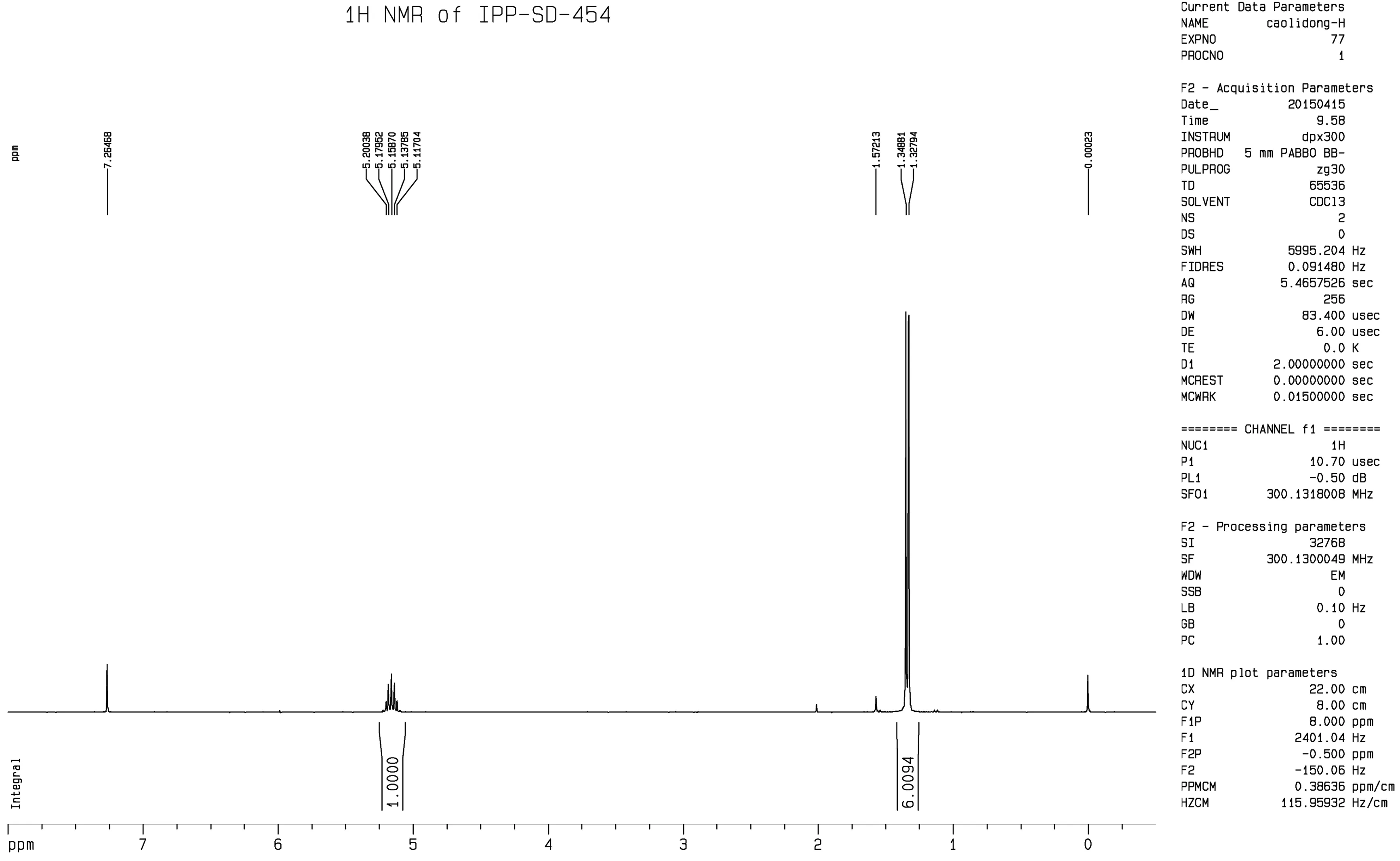
图18 稻瘟灵原药中杂质16 1H NMR谱图
4 结论
原药中杂质制备主要通过定向合成和液相制备色谱富集;杂质纯化主要有柱层析、重结晶、液相制备色谱等三种途径;杂质结构鉴定主要通过核磁、红外、质谱和高分辨质谱进行。以上介绍的副反应类型仅仅是笔者在全分析工作过程中经常遇到的反应类型,实际上原药的品种繁多,可能涉及的副反应类型更多,杂质结构剖析也会遇到更大的挑战。通过上述介绍不难看出,原药中杂质结构剖析是一项难度很大的工作,不仅需要分析化学的技术,更需要具备丰富的有机化学的反应理论和有机合成的实践经验。
猜你喜欢
云南农业科技(2022年1期)2022-01-27
今日农业(2021年2期)2021-11-27
化工管理(2021年7期)2021-05-13
太原师范学院学报(自然科学版)(2021年1期)2021-03-06
今日农业(2020年23期)2020-12-31
含能材料(2020年7期)2020-07-11
今日农药(2017年7期)2017-08-09
今日农药(2014年12期)2015-03-30
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
